دیستروفی قرنیه ژلاتینی قطرهای (Gelatinous Drop-like Corneal Dystrophy: GDLD) یک بیماری ارثی قرنیه است که در آن آمیلوئید در زیر اپیتلیوم قرنیه رسوب کرده و باعث کاهش قابل توجه بینایی دوطرفه میشود.
این بیماری اولین بار در سال 1914 توسط ناکایزومی گزارش شد و در سال 1932 توسط کیوزاوا به عنوان “دژنراسیون قرنیه ژلاتینی قطرهای” نامگذاری شد و از آن زمان با این نام شناخته میشود. در طبقهبندی IC3D (کمیته بینالمللی طبقهبندی دیستروفیهای قرنیه)، در گروه دیستروفیهای اپیتلیال طبقهبندی میشود و مخفف آن GDLD است.
ژن مسئول در سال 1999 توسط تسوجیکاوا و همکاران به عنوان ژن TACSTD2 (ترانسدیوسر سیگنال کلسیم مرتبط با تومور 2) شناسایی شد که یک ژن تک اگزونی واقع در کروموزوم 1p32 است4).
شیوع: در سطح جهانی نادر است، اما در ژاپن به عنوان یک بیماری نسبتاً شایع گزارش شده است 1). تصور میشود با کاهش ازدواجهای فامیلی، فراوانی بروز آن کاهش یافته است 2)
تفاوت منطقهای: در ژاپن نسبتاً شایع است و در اروپا و آمریکا تقریباً گزارش نشده است
جهش Q118X: یک جهش بنیانگذار در بیماران ژاپنی است و بیش از 80% کروموزومهای بیماریزا را تشکیل میدهد 2)
سن شروع: اغلب تا دهه دوم زندگی بروز میکند
در سال 2019، به عنوان یک بیماری نادر تعیین شده و مشمول کمک هزینه پزشکی شد 2). معیارهای تشخیصی و طبقهبندی شدت توسط پروژه تحقیقاتی سیاستگذاری بیماریهای صعبالعلاج وزارت بهداشت، کار و رفاه تهیه شده است 2).
Qآیا GDLD در خارج از ژاپن نیز بروز میکند؟
A
GDLD در سراسر جهان گزارش شده است، اما در ژاپن نسبتاً شایع است. در اروپا و آمریکا تقریباً هیچ موردی از بروز وجود ندارد. بیش از 20 جهش در ژن TACSTD2 گزارش شده است که ناهمگنی ژنتیکی را نشان میدهد. در ژاپن، جهش Q118X واقع در 1p32 به عنوان یک جهش بنیانگذار با فراوانی بالا دیده میشود و گفته میشود بیش از 80% کروموزومهای بیماریزای بیماران ژاپنی را تشکیل میدهد.
اغلب تا دهه دوم زندگی بروز میکند. از دوران کودکی، علائم زیر گزارش میشود.
فوتوفوبی (نورگریزی): یک علامت برجسته از مراحل اولیه است
احساس جسم خارجی: ناشی از برجستگیهای ژلاتینی روی سطح قرنیه
اشکریزش: همراه با علائم تحریکی
کاهش بینایی: با پیشرفت رسوب آمیلوئید به تدریج بدتر میشود. پس از بزرگسالی قابل توجه است
با افزایش سن، تعداد و اندازه رسوبات آمیلوئید افزایش مییابد. این رسوبات به رنگ خاکستری مایل به سفید تا زرد تبدیل میشوند و در نهایت بیشتر قرنیه را به ویژه در ناحیه شکاف پلکی میپوشانند 2). تهاجم عروقی از محیط، کاهش قابل توجه بینایی و درد چشم ایجاد میشود و همراه با مشکلات زیبایی، کیفیت زندگی بیمار را به شدت کاهش میدهد.
یافتههای بالینی (یافتههایی که پزشک در معاینه تأیید میکند)
بین شکاف پلکی: کدورت در لایههای سطحی مشاهده میشود. یافتههایی مشابه کراتوپاتی نواری را نشان میدهد.
ضایعات ملتحمه: ممکن است ضایعاتی در ملتحمه نیز دیده شود.
شکل کامکوات
نوع کامکوات مانند: در موارد پیشرفته شایعتر است.
رسوبات منتشر زرد مایل به سفید: کل قرنیه زرد شده و ظاهری شبیه کامکوات پیدا میکند.
تهاجم عروقی: ممکن است با عروق جدید سطحی همراه باشد.
نوع کدورت استرومایی
نوع کدورت استرومایی (stromal opacity type): مرحله پیشرفتهتر.
گسترش به استروما: ضایعه به استرومای قرنیه میرسد.
تهاجم عروقی: برجستگیهای ژلاتینی به رنگ سفید شیری تا زرد همراه با تهاجم عروقی.
Ide و همکاران طیف بالینی دقیق ۳۴ مورد در ژاپن را گزارش کردند و نشان دادند که حتی با همان جهش ژن TACSTD2 (هوموزیگوت Q118X) چهار فنوتیپ مختلف وجود دارد3).
سایر یافتههای مشخصه عبارتند از:
رنگآمیزی تأخیری با فلورسئین (delayed staining): با وجود عدم آسیب اپیتلیال قرنیه، به دلیل افزایش نفوذپذیری ناشی از نقص اتصالات محکم، فلورسانس چند دقیقه پس از قطره فلورسئین مشاهده میشود2)
نازک شدن اپیتلیوم: در نواحی دارای برجستگیهای ژلاتینی، اپیتلیوم قرنیه نازک میشود
تهاجم عروقی: تهاجم عروقی سطحی در محیط قرنیه مشاهده میشود
به دلیل ناهنجاری در ژن TACSTD2، محلیسازی درون سلولی طبیعی کلودین ۱ و کلودین ۷ که پروتئینهای سازنده اتصالات محکم در اپیتلیوم قرنیه هستند، از بین میرود و عملکرد سد اپیتلیال کاهش مییابد5). در نتیجه، پروتئینهایی مانند لاکتوفرین از اشک وارد قرنیه شده، فیبریلهای آمیلوئید تشکیل داده و در زیر اپیتلیوم رسوب میکنند. ناکاتسوکا و همکاران با تحلیل خانوادههای ژاپنی نشان دادند که TACSTD2 برای محلیسازی طبیعی کلودین ضروری است و پاتوفیزیولوژی GDLD ناشی از اختلال عملکرد اتصالات محکم است5).
سابقه خانوادگی: به دلیل وراثت اتوزومال مغلوب، در صورت ناقل بودن هر دو والد، خطر ابتلا وجود دارد
نژاد ژاپنی: جهش بیمعنی Q118X به عنوان جهش بنیانگذار در ژاپن بیش از 80% کروموزومهای بیماریزا را تشکیل میدهد2)
ازدواج فامیلی: معمولاً والدین فرد مبتلا ازدواج فامیلی دارند. با این حال، افراد با هتروزیگوسیتی مرکب ناشی از ازدواج بین خانوادههای مختلف نیز مبتلا میشوند
Yang Jing, Chun Liu, Liya Wang A novel TACSTD2 mutation identified in two Chinese brothers with gelatinous drop-like corneal dystrophy 2009 Aug 14 Mol Vis. 2009 Aug 14; 15:1580-1588 Figure 5. PMCID: PMC2728569. License: CC BY.
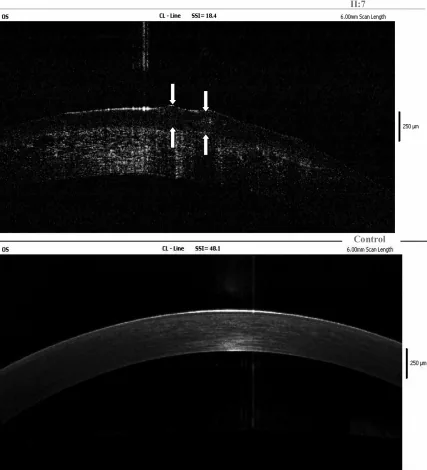
یافتههای OCT دامنه فوریه از قرنیه چشم چپ فرد مبتلا، رسوب آمیلوئید را در اپیتلیوم قرنیه و لایههای سطحی استروما نشان میدهد و فلشها محل ضایعات قطرهای ژلاتینی را نشان میدهند. این تصویر مربوط به رسوب آمیلوئید است که در بخش «4. روشهای تشخیص و آزمایش» بحث شده است.
معیارهای تشخیصی GDLD توسط گروه تحقیقاتی پروژه تحقیقاتی سیاستهای بیماریهای صعبالعلاج وزارت بهداشت، کار و رفاه «تدوین و ترویج راهنمای بالینی برای بیماریهای نادر بخش قدامی چشم» تدوین شده است2). اگر بر اساس این معیارها به عنوان Definite تشخیص داده شود، بیماری نادر مشخص شده محسوب میشود.
A. علائم (هر یک از موارد زیر)
کاهش بینایی
فوتوفوبی (نورگریزی)
احساس جسم خارجی
اشکریزش
B. یافتههای آزمایشگاهی
تجمع رسوبات آمیلوئید به رنگ سفید مایل به خاکستری و برجسته در زیر اپیتلیوم قرنیه در مرکز قرنیه و ناحیه شکاف پلک (شکل توتمانند) در هر دو چشم مشاهده میشود.
با وجود عدم وجود آسیب اپیتلیال قرنیه، رنگآمیزی تاخیری (delayed staining) مشاهده میشود که در آن فلورسانس چند دقیقه پس از رنگآمیزی با فلورسئین قابل مشاهده است.
تهاجم عروق سطحی در قسمت محیطی قرنیه مشاهده میشود.
C. تشخیص افتراقی: آمیلوئیدوز ثانویه قرنیه و کراتوپاتی قطرهای اقلیمی (climatic droplet keratopathy) باید رد شوند.
D. عوارض خارج چشمی: هیچ.
E. آزمایش ژنتیکی: ناهنجاری در ژن TACSTD2 مشاهده میشود.
شرایط قطعی (Definite) زمانی است که یکی از موارد زیر برآورده شود2).
موردی که D را برآورده کند، یکی از موارد A را نشان دهد، B1 را نشان دهد، و بیماریهای قابل تشخیص افتراقی در C را بتوان رد کرد.
موردی که D را برآورده کند، یکی از موارد A را نشان دهد، B2 یا B3 را نشان دهد، E را نشان دهد، و بیماریهای قابل تشخیص افتراقی در C را بتوان رد کرد.
B1 (رسوبات توتمانند) یک یافته بسیار مشخص است و در موارد معمول تشخیص دشوار نیست. در موارد غیرمعمول، تشخیص با ترکیب A تا C و آزمایش ژنتیکی (E) انجام میشود2).
شدت بیماری بر اساس بهترین دید اصلاحشده چشم بهتر به چهار درجه I تا IV طبقهبندی میشود2).
شدت
معیار
کمک هزینه پزشکی
درجه I
فقط یک چشم مبتلا است، چشم دیگر سالم
×
درجه II
هر دو چشم مبتلا، حدت بینایی اصلاحشده چشم بهتر 0.3 یا بیشتر
×
درجه III
هر دو چشم مبتلا، حدت بینایی اصلاحشده چشم بهتر 0.1 یا بیشتر اما کمتر از 0.3
○
درجه IV
هر دو چشم مبتلا، حدت بینایی اصلاحشده چشم بهتر کمتر از 0.1
○
هنگامی که تشخیص Definite داده شود، بیماری نادر مشخص شده محسوب میشود و در صورت شدت درجه III یا بالاتر، میتوان از کمک هزینه پزشکی برخوردار شد2). اگر در چشم بهتر به دلیل گلوکوم ثانویه و غیره تنگی میدان بینایی (میدان بینایی باقیمانده مرکزی 20 درجه یا کمتر با هدف Goldmann I/4) وجود داشته باشد، شدت یک درجه افزایش مییابد.
معاینه با لامپ شکاف: ضایعات برجسته خاکستری مایل به سفید از مرکز قرنیه تا ناحیه شکاف پلک مشاهده میشود و چهار نوع (توتمانند، نواری، کامکواتمانند و کدورت استرومایی) تشخیص داده میشود.
آزمون نفوذ فلورسئین (رنگپذیری تأخیری): به دلیل نارسایی اتصالات محکم، رنگ به سرعت به بافت قرنیه نفوذ میکند2)
توموگرافی انسجام نوری بخش قدامی (AS-OCT): امکان ارزیابی غیرتهاجمی عمق و گستردگی رسوب آمیلوئید در سراسر لایه زیراپیتلیال و استرومای قرنیه را فراهم میکند
آزمایش ژن TACSTD2: از سال 2020 به عنوان آزمایش ژنتیک دیستروفی قرنیه (D006-20) تحت پوشش بیمه قرار گرفته است2). TACSTD2 یک ژن تک اگزونی است و جستجوی آن آسان است. به ویژه برای تشخیص موارد غیر معمول مفید است
بررسی بافتی (مقطع قرنیه): با رنگآمیزی کنگو رد به رنگ نارنجی-قرمز رنگ میشود و در زیر میکروسکوپ پلاریزاسیون دوشکستی سبز سیبی نشان میدهد که آمیلوئید را تأیید میکند
آمیلوئیدوز ثانویه قرنیه: در اثر تحریک مزمن مانند مژههای ناهنجار، انتروپیون، برجستگی قوز قرنیه، یا استفاده از لنزهای تماسی سخت، آمیلوئید رسوب میکند. عدم وجود سابقه خانوادگی و وجود التهاب مزمن سطح چشم از نکات افتراقی است. ممکن است ظاهری شبیه برجستگی ژلاتینی یا شبکهای داشته باشد و برای تأیید نیاز به بررسی بافتی است
کراتوپاتی قطرهای اقلیمی: بیشتر در مردان بالای 40 سال دیده میشود. در مناطق بیابانی یا بسیار سرد رخ میدهد و علت آن اشعه فرابنفش و خشکی است. ضایعات قرنیهای برجسته به رنگ زرد تا خاکستری مایل به سفید ایجاد میکند
دژنراسیون نواری قرنیه: نمکهای کلسیم در زیر اپیتلیوم رسوب میکنند. از ناحیه محیطی در ساعت 3 و 9 شروع شده و به سمت مرکز پیشرفت میکند
دیستروفی شبکهای قرنیه نوع I: ناشی از جهش R124C در ژن TGFBI با وراثت اتوزومال غالب. کدورتهای رشتهای منشعب در استرومای قرنیه ایجاد میکند
دیستروفی لکهای قرنیه: ناشی از ناهنجاری ژن CHST6 با وراثت اتوزومال مغلوب. کدورت منتشر شیشهای مات
Qآیا آزمایش ژنتیک تحت پوشش بیمه انجام میشود؟
A
آزمایش ژن TACSTD2 از سال 2020 به عنوان «آزمایش ژنتیک دیستروفی قرنیه (D006-20)» تحت پوشش بیمه قرار گرفته است. با این حال، لازم است مرکز درمانی توانایی انجام آزمایش را داشته باشد و تأییدیه مرکز را دریافت کند. TACSTD2 یک ژن تک اگزونی است و جستجوی آن آسان است و بیش از 80٪ از بیماران ژاپنی دارای جهش بنیانگذار Q118X هستند، بنابراین این آزمایش به ویژه برای تشخیص موارد غیر معمول مفید است2).
درمان GDLD بر اساس وسعت کدورت و میزان اختلال بینایی انتخاب میشود. از آنجا که این یک بیماری ارثی است، چالش اصلی نرخ بسیار بالای عود با هر روش درمانی است2). مواردی که به دلیل عوارض پیوند مکرر قرنیه و گلوکوم ثانویه به نابینایی منجر میشوند، غیر معمول نیستند.
اشک مصنوعی: به عنوان درمان علامتی برای تسکین تحریک سطحی استفاده میشود
استفاده مداوم از لنزهای تماسی نرم درمانی (SCL): میتواند از عود ضایعات برجسته ژلاتینی جلوگیری کرده و فاصله بین جراحیها را افزایش دهد
استفاده مداوم از لنزهای تماسی نرم درمانی به عنوان یک درمان محافظهکارانه و کمکی در نظر گرفته میشود 6). Maeno و همکاران در سال 2020 در یک مطالعه مشاهدهای آیندهنگر بر روی بیماران GDLD نشان دادند که استفاده از لنزهای تماسی درمانی به طور قابل توجهی از عود ضایعات برجسته ژلاتینی خاکستری-سفید تا زرد جلوگیری میکند 7). همچنین برای پیشگیری از عود پس از جراحی توصیه میشود.
کراتکتومی سطحی درمانی با لیزر اگزایمر: انتخاب اول برای کدورتهای سطحی.
اندیکاسیون: برجستگیهای ژلاتینی سطحی خفیف تا متوسط. همراه با خراش دستی. نتایج بلندمدت 8,9).
پیوند قرنیه
لایهای، لایهای عمیق (DALK) و تمام ضخامت (PKP): برای موارد پیشرفته اندیکاسیون دارد.
میزان عود: عود پس از پیوند تمام ضخامت در عرض 4 سال 97% است که بالا میباشد. در DALK مزیت حفظ اندوتلیوم وجود دارد.
پیوند لیمبوس قرنیه
پیوند سلولهای بنیادی لیمبوس و کراتوپلاستی اپیتلیال: همراه با پیوند قرنیه استفاده میشود.
هدف: پوشش سطح چشم با اپیتلیوم قرنیه مشتق از پیوند و جلوگیری از نفوذ مجدد اپیتلیوم میزبان 10,11).
نتایج بلندمدت PTK از چندین گزارش در ژاپن منتشر شده است. Ōura و همکاران نتایج بلندمدت PTK را برای موارد GDLD نشان دادند و مفید بودن آن را در افزایش فاصله تا عود گزارش کردند 8). Hieda و همکاران در یک مطالعه چندمرکزی در ژاپن زمان عود و پیامدهای بالینی پس از PTK را به طور دقیق تجزیه و تحلیل کردند 9).
استفاده ترکیبی از پیوند سلولهای بنیادی لیمبوس (LSCT) به عنوان یک رویکرد ژاپنی در سطح جهانی مورد ارزیابی قرار گرفته است. Shimazaki و همکاران در سال 2002 اثربخشی پیوند قرنیه همراه با LSCT را برای GDLD گزارش کردند و نشان دادند که با جلوگیری از نفوذ مجدد سلولهای اپیتلیال میزبان میتوان فاصله تا عود را افزایش داد 10). پس از آن Movahedan و همکاران نیز رویکرد مشابهی را گزارش کردند 11).
برای پوشاندن سطح چشم با اپیتلیوم قرنیه مشتق از پیوند، ابتدا اپیتلیوم قرنیه میزبان برداشته شده و سپس پیوند لیمبوس انجام میشود. پس از عمل، استفاده مداوم از لنز تماسی درمانی ادامه یافته و عود بیماری به تأخیر میافتد.
در سالهای اخیر، استفاده از قرنیه مصنوعی (Boston type I Kpro) نیز مورد بررسی قرار گرفته است. از آنجایی که این روش به اپیتلیوم قرنیه میزبان نیاز ندارد، از نظر تئوری از رسوب مجدد آمیلوئید جلوگیری میکند، اما خطر عوارض پس از عمل مانند عفونت و غشای پشت قرنیه مصنوعی وجود دارد.
Qآیا پس از پیوند قرنیه نیز بیماری عود میکند؟
A
در GDLD، میزان عود پس از پیوند قرنیه بسیار بالاست. گزارش شده است که حدود 97٪ موارد در عرض 4 سال پس از پیوند نافذ قرنیه (PKP) عود میکنند. علت اصلی جایگزینی سلولهای اپیتلیال گیرنده با اپیتلیوم پیوند است. به عنوان یک راهکار ژاپنی، استفاده همزمان از پیوند سلولهای بنیادی لیمبوسقرنیه10) و استفاده مداوم از لنز تماسی درمانی 7) برای تأخیر در عود انجام میشود. در مدیریت طولانیمدت، با فرض عود، هدف حفظ عملکرد بینایی و افزایش فاصله بین جراحیها است.
ژن TACSTD2 که عامل GDLD است، یک ژن تک اگزونی واقع در بازوی کوتاه کروموزوم 1 (1p32) میباشد. در سال 1999، تسوجیکاوا و همکاران با آنالیز پیوند در خانوادههای ژاپنی آن را به عنوان ژن عامل شناسایی کردند 4). پروتئین TACSTD2 نقش ضروری در حفظ عملکرد سد اپیتلیال قرنیه ایفا میکند.
جهشهای از دستدهنده عملکرد در ژن TACSTD2 باعث اختلال در جایگاهیابی طبیعی درون سلولی پروتئینهای Claudin 1 و Claudin 7 که اجزای اتصالات محکم هستند، میشود. ناکاتسوکا و همکاران با استفاده از سلولهای اپیتلیال قرنیه کشتشده و آنالیز خانوادههای ژاپنی نشان دادند که از دست دادن عملکرد TACSTD2 منجر به از دست رفتن جایگاهیابی Claudin در محل اتصالات جانبی-راسی و کاهش عملکرد سد اپیتلیال میشود 5). علاوه بر این، در سال 2011، سه جهش جدید TACSTD2 و جایگاهیابی غیرطبیعی درون سلولی آنها را گزارش کردند 12).
کاهش عملکرد سد اپیتلیال باعث نفوذ پروتئینهایی مانند لاکتوفرین از اشک به داخل قرنیه میشود. لاکتوفرین نفوذ کرده، فیبریلهای آمیلوئید را تشکیل داده و در زیر اپیتلیوم قرنیه رسوب میکند. رسوبات آمیلوئید حاوی لاکتوفرین هستند، اما این بیماری ناشی از ناهنجاری در ژن لاکتوفرین نیست.
از نظر بافتشناسی، کدورت شیریرنگ زیر اپیتلیوم با رنگآمیزی کنگو رد به رنگ نارنجی-قرمز دیده میشود و در میکروسکوپ پلاریزاسیون، دوشکستی سبز سیب (apple-green) نشان میدهد. در میکروسکوپ الکترونی، اتصالات محکم اپیتلیوم با فضاهای شفاف الکترونی جایگزین شدهاند. رسوبات به داخل لاملهای قرنیه نیز نفوذ کرده و باعث تخریب فیبرهای کلاژن و پروتئوگلیکانها میشوند.
بیش از ۲۰ جهش در ژن TACSTD2 گزارش شده است12). در ژاپن، جهش Q118X (جهش بیمعنی، فانکشنال نال) به عنوان جهش بنیانگذار بیش از ۸۰% کروموزومهای بیماریزا را تشکیل میدهد2). بیماری معمولاً در حالت هموزیگوت بروز میکند، اما در هتروزیگوتهای مرکب ناشی از ازدواج بین خانوادههای مختلف نیز دیده میشود. جالب توجه است که حتی در افراد با جهش هموزیگوت Q118X یکسان، چهار نوع بالینی توتتوتمانند، نواری، کامکواتمانند و کدورت پارانشیمی به صورت مخلوط مشاهده میشود3).
آمیلوئیدوز قرنیه بر اساس اولیه یا ثانویه بودن و سیستمیک یا موضعی بودن طبقهبندی میشود. GDLD همراه با دیستروفی قرنیه مشبک در گروه آمیلوئیدوز اولیه موضعی قرار میگیرد. دژنراسیون آمیلوئیدی ثانویه موضعی به دنبال تریکیازیس، قوز قرنیه، تروما، استفاده طولانی مدت از لنز تماسی و غیره ایجاد میشود و باید در تشخیص افتراقی در نظر گرفته شود.
GDLD یک بیماری نادر است و در هر مرکز پزشکی، پزشکان با تجربه بالینی کم هستند و روشهای تشخیصی و درمانی استاندارد وجود نداشت. توسط گروه تحقیقاتی اپیدمیولوژی بیماریهای نادر قرنیه (پروژه تحقیقاتی سیاستگذاری بیماریهای صعبالعلاج وزارت بهداشت، کار و رفاه) و گروه تحقیقاتی تدوین و ترویج راهنمای بالینی بیماریهای نادر بخش قدامی چشم، معیارهای تشخیصی و طبقهبندی شدت بیماری تدوین شد2). در سال ۲۰۱۹، این بیماری به عنوان بیماری نادر تعیین شده «دیستروفی قرنیه قطرهای ژلاتینی» ثبت شد و در حال حاضر راهنمای بالینی مبتنی بر Minds (سرویس توزیع اطلاعات پزشکی) در حال تدوین است2).
در بلندمدت، عود و درمانهای مکرر مشکلساز هستند. استفاده همزمان از لنز تماسی درمانی و پیوند لیمبوس میتواند عود را به تأخیر انداخته و فاصله بین عملها را افزایش دهد و به بهبود پیشآگهی طولانی مدت کمک کند6, 7, 10).
Maeno و همکاران یک مورد بالینی غیر معمول با رسوب آمیلوئید عودکننده تنها در یک چشم گزارش کردند که تنوع فنوتیپی GDLD را نشان میدهد13). در تشخیص چنین موارد غیر معمول، آزمایش ژنتیکی TACSTD2 نقش تعیینکنندهای دارد2).
در تحقیقات پایهای، انتظار میرود که دینامیک مولکولهای اتصال محکم در پاییندست TACSTD2 به طور دقیق روشن شود و درمانهایی با هدف تثبیت کلودین توسعه یابد. از نظر بالینی، گسترش اندیکاسیون رویکردهای پزشکی بازساختی مانند قرنیه مصنوعی، پیوند لایه اپیتلیال قرنیه و اپیتلیوم قرنیه مشتق از سلولهای iPS در حال بررسی است.
Ide T, Nishida K, Maeda N, Tsujikawa M, Yamamoto S, Watanabe H, et al. A spectrum of clinical manifestations of gelatinous drop-like corneal dystrophy in japan. American journal of ophthalmology. 2004;137(6):1081-4. doi:10.1016/j.ajo.2004.01.048. PMID:15183793.
Tsujikawa M, Kurahashi H, Tanaka T, Nishida K, Shimomura Y, Tano Y, et al. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nature genetics. 1999;21(4):420-3. doi:10.1038/7759. PMID:10192395.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Tsujikawa M, et al. Tumor-associated calcium signal transducer 2 is required for the proper subcellular localization of claudin 1 and 7: implications in the pathogenesis of gelatinous drop-like corneal dystrophy. Am J Pathol. 2010;177(3):1344-1355. PMID: 20651236. PMCID: PMC2928967. doi:10.2353/ajpath.2010.100149.
Kawasaki S, Kinoshita S. Clinical and basic aspects of gelatinous drop-like corneal dystrophy. Dev Ophthalmol. 2011;48:97-115. PMID: 21540633. doi:10.1159/000324079.
Maeno S, Soma T, Tsujikawa M, Shigeta R, Kawasaki R, Oie Y, et al. Efficacy of therapeutic soft contact lens in the management of gelatinous drop-like corneal dystrophy. The British journal of ophthalmology. 2020;104(2):241-246. doi:10.1136/bjophthalmol-2018-313809. PMID:31023713.
Hieda O, Kawasaki S, Yamamura K, Nakatsukasa M, Kinoshita S, Sotozono C. Clinical outcomes and time to recurrence of phototherapeutic keratectomy in Japan. Medicine. 2019;98(27):e16216. doi:10.1097/MD.0000000000016216. PMID:31277131; PMCID:PMC6635226.
Shimazaki J, Shimmura S, Tsubota K. Limbal stem cell transplantation for the treatment of subepithelial amyloidosis of the cornea (gelatinous drop-like dystrophy). Cornea. 2002;21(2):177-80. doi:10.1097/00003226-200203000-00010. PMID:11862090.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Nishida K, et al. Two novel mutations of TACSTD2 found in three Japanese gelatinous drop-like corneal dystrophy families with their aberrant subcellular localization. Molecular vision. 2011;17:965-70. PMID:21541270; PMCID:PMC3084224.
Maeno S, Soma T, Nishida K. A Case of Clinically Atypical Gelatinous Drop-like Corneal Dystrophy With Unilateral Recurrent Amyloid Depositions. Cornea. 2022;41(11):1447-1450. doi:10.1097/ICO.0000000000003070. PMID:36219213.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.