دیستروفی شبکهای قرنیه (lattice corneal dystrophy, LCD) یک دیستروفی ارثی قرنیه است که در آن آمیلوئید در استرومای قرنیه رسوب کرده و کدورتهای خطی شبکهای ایجاد میکند. این بیماری سابقه طولانی دارد و از دهه 1890 توصیف شده است. در طبقهبندی بالینی-ژنتیکی ویرایش دوم IC3D (کمیته بینالمللی طبقهبندی دیستروفیهای قرنیه) به LCD1 و واریانتهای آن (انواع 3، 3A، 1/3A و 4 سابق) ادغام شده است4).
LCD1، دیستروفی گرانولار قرنیه، دیستروفی قرنیه Reis-Bücklers و دیستروفی قرنیه Thiel-Behnke گروهی از بیماریها را به عنوان «دیستروفیهای مرتبط با TGFBI» تشکیل میدهند. ژن عامل TGFBI (ژن القا شده توسط فاکتور رشد تبدیلکننده بتا) روی بازوی بلند کروموزوم 5 (5q31) قرار دارد و به صورت اتوزومال غالب به ارث میرسد. پروتئین TGFBI (TGFBIp، کراتواپیتلین، βig-h3) توسط اپیتلیوم قرنیه تولید شده و در تمام لایههای قرنیه توزیع میشود. در استرومای قرنیه در ساختار فیبرهای کلاژن نقش دارد. حتی با جهش در یک ژن، تفاوت در محل جهش و اسید آمینه جایگزین منجر به تمایز زیاد در ماده رسوبی (هیالین یا آمیلوئید) و تصویر بالینی میشود5).
جهش اصلی LCD1 R124C است که در آن آرژنین 124 در ژن TGFBI با سیستئین جایگزین میشود. در نوع واریانت LCD IIIA، جهشهایی مانند L527R گزارش شده است.
پروتئین غیرطبیعی تجمعیافته در قرنیه با رنگآمیزی کنگو رد به رنگ قرمز درآمده و در میکروسکوپ پلاریزاسیون دوشکستی سبز سیب مشخصی را نشان میدهد که به عنوان آمیلوئید تأیید میشود. این یافته از قرن نوزدهم یک شاخص تشخیصی بافتی کلاسیک برای آمیلوئیدوز بوده است6).
نوعی که قبلاً «دیستروفی قرنیه مشبک نوع 2» نامیده میشد، تظاهر چشمی آمیلوئیدوز سیستمیک ژلسولینی (GSN-AMYL، سندرم مرتویا) است و در طبقهبندی فعلی IC3D به عنوان «آمیلوئیدوز خانوادگی» طبقهبندی شده و جدا از LCD کلاسیک در نظر گرفته میشود4,10). این سندرم که در سال 1969 توسط مرتویا فنلاندی توصیف شد، یک بیماری ارثی است که علاوه بر کدورتهای مشبک قرنیه، با نوروپاتی پیشرونده جمجمهای، شلی پوست و علائم سیستمیک همراه است10,11). از آنجایی که تمایز این دو در عمل بالینی مهم است، در این مقاله هر دو ذکر میشوند.
کدورتهای رشتهای دو حلقهای در ناحیه مردمک، فرسایش مکرر اپیتلیوم
LCD IIIA (نوع واریانت)
TGFBI (5q31)
L527R و غیره
بعد از دهه ۴۰ سالگی
خطوط مشبک ضخیم طنابمانند در لایه عمقی استروما، بدون اختلال اپیتلیال
نوع GSN (Meretoja)
GSN (9q34)
D187N، p.Glu580Lys2)
دهه ۳۰ تا ۴۰ سالگی
خطوط مشبک شعاعی در محیط، آمیلوئیدوز سیستمیک
در ژاپن، دیستروفیهای مرتبط با TGFBI که شایعتر هستند، عمدتاً از نوع گرانولار نوع II (Avellino، R124H) میباشند و LCD1 در مقایسه با آن نادرتر است. با این حال، از آنجایی که هر دو تنها با تفاوت چند نوکلئوتیدی در همان ژن TGFBI از یکدیگر متمایز میشوند، در مواردی که تصویر بالینی همپوشانی دارد، تأیید با آزمایش ژنتیکی توصیه میشود. شیوع دقیق LCD در ژاپن گزارش نشده است، اما در میان کل دیستروفیهای قرنیه نسبتاً نادر محسوب میشود.
Qتفاوت LCD1 و سندرم Meretoja چیست؟
A
LCD1 رسوب آمیلوئید محدود به قرنیه ناشی از جهش در ژن TGFBI است که در دهه دوم تا سوم زندگی از ناحیه مردمک شروع شده و با فرسایش مکرر اپیتلیال همراه است. در مقابل، سندرم Meretoja (LCD2 سابق، نوع GSN) تظاهر چشمی آمیلوئیدوز سیستمیک ناشی از جهش در ژن GSN (ژلسولین) است که در دهه سوم تا چهارم زندگی از قرنیه محیطی شروع شده و شفافیت مرکز قرنیه برای مدت طولانی حفظ میشود. سندرم Meretoja با علائم سیستمیک مانند شلی پوست، صورت ماسکمانند، نوروپاتی محیطی و آریتمی قلبی همراه است2,10). در ویرایش دوم IC3D، سندرم Meretoja به طور مستقل از دیستروفی مشبک قرنیه طبقهبندی میشود4).
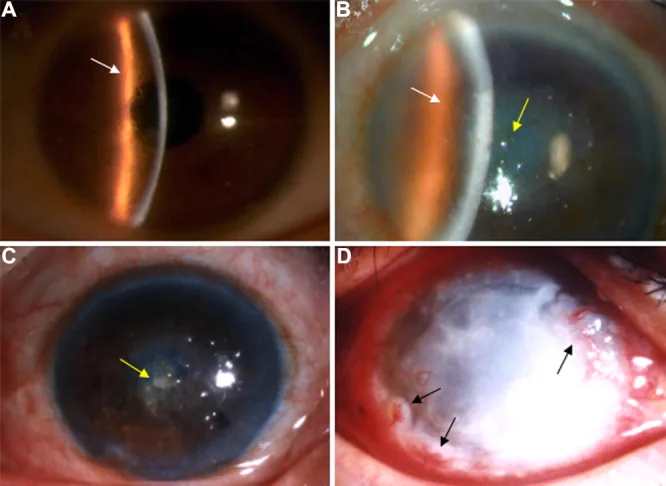
Liu Z, et al. An R124C mutation in TGFBI caused lattice corneal dystrophy type I with a variable phenotype in three Chinese families. Mol Vis. 2008. Figure 2. PMCID: PMC2443752. License: CC BY.
در عکس با لامپ شکافی، خطوط مشبک در استروما و کدورت غالب در مرکز قرنیه دیده میشود. این تصویر یافتههای بالینی مشخصه دیستروفی قرنیه مشبک را نشان میدهد.
در LCD1، بسیاری از بیماران در دوران کودکی بدون علامت هستند و تنها کدورتهای ریز وجود دارد که با روش عبور نور در میکروسکوپ لامپ شکافی قابل تشخیص است. از دهه دوم تا سوم زندگی، فرسایش مکرر اپیتلیوم قرنیه (RCE) رخ میدهد که با درد شدید چشم، فتوفوبی، اشکریزی و احساس جسم خارجی در هنگام بیدار شدن از خواب همراه است. در حدود 30 سالگی، کدورت سفید در لایههای سطحی استرومای مرکزی قرنیه آشکار میشود و پس از 40 سالگی کاهش بینایی پیشرفت میکند.
در LCD IIIA (نوع واریانت)، معمولاً آسیب اپیتلیال رخ نمیدهد و شکایت اصلی کاهش تدریجی بینایی پس از 40 سالگی است.
در LCD2 قدیمی (سندرم Meretoja)، علائم چشمی در دهه سوم تا چهارم زندگی ظاهر میشود، اما اختلال بینایی قابل توجه اغلب تا دهه ششم به تأخیر میافتد11). علائم سیستمیک مانند شلی پوست پلک، صورت ماسکمانند، نوروپاتی پیشرونده کرانیال و آریتمی قلبی اغلب پیش از علائم چشمی یا همزمان با آن ظاهر میشوند2,10).
یافتههای میکروسکوپ لامپ شکافی بر اساس نوع بیماری در زیر ارائه شده است.
LCD1 (نوع کلاسیک)
محل شروع: به صورت کدورتهای ریز نقطهای و خطی در لایه Bowman تا استرومای سطحی در ناحیه مردمک هر دو چشم ظاهر میشود.
خطوط مشبک: کدورتهای رشتهای و خطی با حاشیه دوگانه که در هم تنیده شده و کدورتهای شبکهای و ستارهای شکل را تشکیل میدهند.
مرحله پیشرفته: کدورت سفید شیری به شکل تخممرغی یا دایرهای در مرکز قرنیه ایجاد میشود.
روش نور بازتابی: خطوط مشبک نازک و نیمه شفاف که در نور مستقیم به سختی دیده میشوند، به وضوح نمایان میشوند.
رنگآمیزی فلورسئین: به دلیل کاهش چسبندگی اپیتلیوم، سطح ناهموار میشود.
فرسایش مکرر اپیتلیوم: به دلیل رسوبات در سلولهای پایه اپیتلیوم و غشای Bowman، با فراوانی بالا رخ میدهد.
LCD IIIA (نوع واریانت)
خطوط شبکهای: خطوط شبکهای ضخیم و بلند در لایههای میانی تا عمقی استروما، گاهی با انشعابات درختی. با نور مستقیم نیز قابل مشاهده است.
فنوتیپ: سه الگو وجود دارد: ① فقط خطوط شبکهای، ② فقط رسوبات دانهای کوچک، ③ ترکیبی از هر دو. ممکن است در یک فرد، چشمهای چپ و راست فنوتیپ متفاوتی داشته باشند یا موارد یکطرفه دیده شود.
اپیتلیوم: معمولاً آسیب اپیتلیال رخ نمیدهد.
هوموزیگوت: در هوموزیگوتهای L527R، خطوط شبکهای ضخیمتر و رسوبات دانهای مرکزی بزرگتر هستند، اما تفاوت به اندازه تفاوت بین هتروزیگوت و هوموزیگوت در R124H (نوع دانهای II) بارز نیست.
نوع GSN (مرتویا)
خطوط شبکهای: تعداد کمی رسوب شبکهای که ظرافت ندارند، به صورت شعاعی از محیط ظاهر میشوند.
شفافیت مرکزی: شفافیت ناحیه مرکزی برای مدت طولانی پس از شروع بیماری حفظ میشود.
فرسایش اپیتلیال: نادر است.
یافتههای سیستمیک: تغییرات چهره مانند صورت ماسکمانند، لبهای برجسته همراه با اختلال حرکتی، گوشهای آویزان، و شلی پوست پلک (بلفاروکالازی) دیده میشود2).
در LCD1، در برخی موارد کدورت دایرهای مرکزی به ویژه شدید میشود؛ گزارشی از یک هتروزیگوت R124C 56 ساله وجود دارد که به دلیل کدورت دایرهای مرکزی نیاز به پیوند قرنیه پیدا کرد.
Qآیا میتوان LCD1 را در کودکان تشخیص داد؟
A
LCD1 در دوران کودکی اغلب بدون علامت است و با نور مستقیم به سختی قابل تشخیص است. با استفاده از روشهای دقیق مانند نور عبوری (transillumination) یا نور بازتابی (retroillumination) در میکروسکوپ اسلیت لمپ، میتوان کدورتهای نقطهای تا خطی ریز در لایههای سطحی استروما را مشاهده کرد. در کودکانی که فرسایش مکرر اپیتلیال قرنیه دارند، باید LCD1 را مد نظر داشت و ارزیابی شامل گرفتن سابقه خانوادگی و معاینه قرنیه والدین توصیه میشود. آزمایش ژنتیکی TGFBI برای تشخیص قطعی مفید است.
جهش شایع LCD IIIA: L527R (Leu527Arg) و موارد دیگر گزارش شده است. موارد هموزیگوت نیز وجود دارد.
جهش de novo: جهش de novo TGFBI L509P در بیماری با فنوتیپ LCD IIIA گزارش شده است1). والدین جهش نداشتند و جهش به یکی از فرزندان منتقل شده بود1).
نقش TGFBIp: توسط اپیتلیوم قرنیه تولید شده و در تمام لایههای قرنیه توزیع میشود؛ در استروما در ساختار فیبرهای کلاژن نقش دارد5).
مرتبط با GSN (سندرم مرتویا، LCD2 سابق)
مکان ژنی: 9q34 (ژن GSN، ژلسولین).
الگوی وراثت: اتوزومال غالب.
جهش کلاسیک: D187N (نوع فنلاندی) شایعترین است و p.Asp187Tyr نیز گزارش شده است10,11).
جهش جدید: p.Glu580Lys گزارش شده در یک خانواده اسلوونیایی در مرز دامنه G4-G5 قرار دارد و با جایگزینی بار منفی با بار مثبت باعث دافعه الکترواستاتیک میشود2).
تصویر بالینی: علاوه بر کدورتهای شبکهای قرنیه، با شلی پوست، آریتمی قلبی، نارسایی کلیوی و نوروپاتی بینایی به صورت آمیلوئیدوز سیستمیک همراه است2).
از آنجا که این بیماری ارثی است، سابقه خانوادگی مهمترین عامل خطر است. با این حال، در TGFBI جهشهای de novo نیز ممکن است رخ دهند، بنابراین عدم وجود سابقه خانوادگی به تنهایی نمیتواند بیماری را رد کند1). الگوی وراثت اتوزومال غالب است و اگر یکی از والدین ناقل جهش باشد، احتمال انتقال به فرزند ۵۰٪ است. تفاوت جنسیتی مشاهده نمیشود و تفاوت نژادی در LCD1 واضح نیست، اما سندرم مرتویا به دلیل تجمع خانوادههای مبتلا در فنلاند شناخته شده است11).
نقش عوامل محیطی مشخص نیست و شروع و پیشرفت بیماری اساساً توسط ژنوتیپ تعیین میشود. با این حال، فراوانی فرسایشهای مکرر اپیتلیال ممکن است در محیط خشک، استفاده از لنزهای تماسی یا ضربه افزایش یابد. جراحیهای انکساری (مانند LASIK، SMILE) میتوانند باعث تشدید سریع دیستروفیهای مرتبط با TGFBI شوند و در غربالگری قبل از عمل، موارد با سابقه خانوادگی نیاز به احتیاط دارند5).
برای تمایز بین LCD1 و انواع واریانت و همچنین نوع GSN، باید یافتههای لامپ شکتی، بافتشناسی و ژنتیک را به طور جامع در نظر گرفت.
آزمایشهای بالینی
میکروسکوپ لامپ شکتی: در نور مستقیم، خطوط شبکهای اولیه به راحتی نادیده گرفته میشوند. با روش عبور نور، کدورتهای ریز در برابر مردمک و با روش بازتاب، خطوط شبکهای نازک نیمه شفاف تشخیص داده میشوند.
رنگآمیزی فلورسئین: در LCD1، به دلیل کاهش چسبندگی اپیتلیوم، رنگآمیزی زبر میشود. همچنین برای ارزیابی وسعت فرسایش اپیتلیوم مفید است.
توموگرافی انسجام نوری بخش قدامی (OCT بخش قدامی): میتوان عمق لایهای رسوبات را به صورت کمی ارزیابی کرد. اندازهگیری عمق ضایعه با FD-OCT برای تعیین عمق برداشتن در PTK مفید است1).
میکروسکوپ کانفوکالقرنیه: امکان مشاهده رسوبات درون استروما در سطح سلولی را فراهم میکند.
تشخیص قطعی
آزمایش ژنتیک: تشخیص نوع بیماری با شناسایی جهشهای ژنهای TGFBI و GSN قطعی میشود. حتی با فنوتیپ یکسان، جهشهای مختلف سرعت عود و پیشرفت را تغییر میدهند، بنابراین مستقیماً به برنامه درمانی مرتبط است.
آزمایش بافتشناسی: رنگآمیزی کنگو رد رنگ قرمز ایجاد میکند و در میکروسکوپ پلاریزاسیون دوشکستی سبز سیبی نشان میدهد که آمیلوئید را تأیید میکند6).
ایمونوهیستوشیمی: تمایز نوع بیماری با آنتیبادیهای ضد TGFBIp و ضد ژلسولین امکانپذیر است.
بررسی سابقه خانوادگی: به دلیل وراثت اتوزومال غالب، بررسی یافتههای قرنیه والدین و خواهر و برادر از تشخیص حمایت میکند.
دیستروفی گرانولار قرنیه نوع II (نوع آولینو، TGFBI R124H): شایعترین دیستروفی مرتبط با TGFBI در ژاپن است و ترکیبی از رسوبات گرانولار و خطوط شبکهای را نشان میدهد. برای تمایز از LCD1، آزمایش ژنتیک قطعی است.
آمیلوئیدوز ثانویه قرنیه: غیر ارثی است و آمیلوئید به صورت ثانویه در زمینه تحریک مزمن سطح چشم مانند مژههای ناهنجار یا قوز قرنیه رسوب میکند. عدم وجود سابقه خانوادگی و وجود بیماری زمینهای از نکات افتراقی است.
دیستروفی لکهای قرنیه: یک بیماری اتوزومال مغلوب ناشی از جهش در ژن CHST6 است که با کدورت منتشر شیشهای و ناهنجاری اندوتلیال همراه است.
دیستروفی ژلاتینی قطرهای قرنیه: یک بیماری اتوزومال مغلوب ناشی از جهش در ژن TACSTD2 است که با برجستگیهای ژلاتینی شیری رنگ تظاهر میکند. در ژاپن نسبتاً شایع است.
Qچرا آزمایش ژنتیک مهم است؟
A
در دیستروفی قرنیه مشبک، حتی اگر فنوتیپ مشابه باشد، تفاوت در ژن عامل و محل جهش باعث تغییرات عمده در سرعت پیشرفت، فراوانی عود، انتخاب درمان و وجود عوارض سیستمیک میشود. LCD1 ناشی از جهش TGFBI و سندرم Meretoja ناشی از جهش GSN از نظر استراتژی درمان و نیاز به بررسی سیستمیک کاملاً متفاوت هستند2,10). علاوه بر این، مواردی از جهشهای de novo گزارش شده است که در آنها سابقه خانوادگی به تنهایی برای تعیین نوع بیماری کافی نیست1)، بنابراین آزمایش ژنتیک برای تشخیص قطعی و طبقهبندی نوع بیماری ضروری است.
در دوران کودکی تا جوانی که بیمار بدون علامت است یا فقط کدورتهای ریز دارد، پیگیری انجام میشود. پیشرفت بیماری با معاینه با لامپ شکاف هر ۶ ماه تا یک سال ارزیابی میشود.
برای فرسایش مکرر اپیتلیوم قرنیه که علامت اصلی LCD1 است، درمان محافظهکارانه زیر مرحله اول است.
درمان حمله: استفاده مداوم از لنزهای تماسی نرم درمانی برای محافظت از اپیتلیوم قرنیه. استفاده همزمان از قطرههای آنتیبیوتیک برای پیشگیری از عفونت ثانویه. استفاده از پماد چشمی برای روانسازی و محافظت از اپیتلیوم.
پیشگیری از عود: استفاده از پماد چشمی قبل از خواب میتواند از عود حملات RCE جلوگیری کند. در محیطهای خشک، استفاده از اشک مصنوعی یا روانکنندهها در طول روز توصیه میشود.
در LCD1 که عمدتاً رسوب آمیلوئید در لایههای سطحی قرنیه است، در موارد کدورت مرکزی شدید یا فرسایش مکرر اپیتلیوم قرنیه، کراتکتومی فوتوتراپی (PTK) با استفاده از لیزر اگزایمر درمان خط اول است7,8). معمولاً عود زودرس رخ نمیدهد، اما عود در طول زمان اجتنابناپذیر است و درمان PTK را میتوان تا حدود دو بار در همان چشم انجام داد.
در افراد هتروزیگوت، عود کند است و موارد نیازمند درمان مجدد نادر است. در افراد هموزیگوت، تمایل به عود زودتر نسبت به هتروزیگوتها وجود دارد. میزان عود پس از PTK مشابه سایر دیستروفیهای مرتبط با TGFBI با گذشت زمان افزایش مییابد و در پیگیری طولانیمدت، در بسیاری از موارد نشانههایی از عود مشاهده میشود8).
به عنوان نمونهای از اثربخشی PTK، در یک مورد LCD IIIA ناشی از جهش de novo TGFBI L509P، PTK به عمق 60 میکرومتر با هدایت FD-OCT انجام شد و بهترین corrected visual acuity (BCVA) از 20/400 به 20/50 بهبود یافت1). در 45 ماه پس از عمل، کاهش بینایی یا عود قابل توجهی مشاهده نشد1).
طبق Preferred Practice Pattern ادم و کدورت قرنیه AAO، PTK برای دیستروفیهای گرانولار و لاتیس قرنیه یک «گزینه منطقی» است و میتواند نیاز به DALK یا پیوند تمام ضخامت قرنیه را به تأخیر بیندازد، اما خطر کدورت پس از عمل وجود دارد. در موارد تکراری، استفاده از میتومایسین C به عنوان روشی برای مهار اسکار عودکننده و رسوبات استرومایی بررسی میشود و هشدار داده میشود که اگر ابلاسیون بیش از یک سوم قدامی استروما باشد یا ضخامت بستر باقیمانده کمتر از 250 میکرومتر باشد، خطر اکتازی قرنیه افزایش مییابد7).
در موارد عود مکرر یا کدورت گسترده به لایههای عمیقتر از استرومای میانی، پیوند قرنیه انتخاب میشود. در LCD1 معمولاً تا پس از 40 سالگی نیاز به پیوند قرنیه ایجاد نمیشود. در LCD سلولهای اندوتلیال قرنیه معمولاً طبیعی هستند، بنابراین روش جراحی بر اساس عمق کدورت انتخاب میشود.
در سالهای اخیر، به دلیل کاهش خطر رد پیوند و نتایج بینایی قابل مقایسه با پیوند تماملایه قرنیه، DALK به عنوان گزینه اول جدید به طور گسترده استفاده میشود.
عود LCD پس از پیوند قرنیه پدیدهای اجتنابناپذیر است و میزان عود پس از پیوند تماملایه قرنیه در ۵ سال ۱۷.۸٪، در ۸ سال ۲۶٪ و در ۱۵ سال ۵۶٪ گزارش شده است9). از آنجایی که کدورت عود معمولاً به لایههای سطحی محدود میشود، میتوان آن را با PTK برداشت و فاصله تا پیوند مجدد را افزایش داد. در مورد LCD IIIA (نوع واریانت)، اغلب تا زمانی که تأثیر شدیدی بر بینایی نداشته باشد، نیازی به درمان نیست.
QPTK تا چه حد مؤثر است؟
A
PTK میتواند رسوبات آمیلوئید سطحی را به طور مؤثر حذف کرده و بهبود بینایی و کاهش فرسایشهای مکرر اپیتلیال را به همراه داشته باشد. در یک مورد LCD IIIA، پس از PTK به عمق ۶۰ میکرومتر، بهترین حدت بینایی اصلاح شده از ۲۰/۴۰۰ به ۲۰/۵۰ بهبود یافت و به مدت ۴۵ ماه عودی مشاهده نشد1). در هتروزیگوتها عود کند است، اما در هموزیگوتها عود زودرس دیده میشود. ضایعات عمیق با PTK قابل برداشتن نیستند، بنابراین برای کدورتهای عمیق نیاز به DALK یا پیوند تماملایه قرنیه است7).
Qآیا پس از پیوند قرنیه عود رخ میدهد؟
A
عود LCD پس از پیوند قرنیه اجتنابناپذیر است. میزان عود پس از پیوند تماملایه قرنیه در ۵ سال ۱۷.۸٪، در ۸ سال ۲۶٪ و در ۱۵ سال ۵۶٪ گزارش شده است9). با این حال، کدورت عود معمولاً به لایههای سطحی پیوند محدود میشود، بنابراین میتوان آن را با PTK برداشت و عمر پیوند را افزایش داد. پیوند لایهای عمیق قرنیه (DALK) در مقایسه با پیوند تماملایه قرنیه خطر رد پیوند اندوتلیال کمتری دارد و به عنوان گزینه اول جدید مورد توجه قرار گرفته است7).
مرکز پاتولوژی LCD1 تجمع غیرطبیعی TGFBIp (کراتو-اپیتلین، βig-h3) است. TGFBIp به طور طبیعی توسط اپیتلیوم قرنیه تولید شده و در تمام لایههای قرنیه توزیع میشود و در استروما به عنوان یک پروتئین ساختاری در ساخت فیبرهای کلاژن و چسبندگی سلولی نقش دارد5). پروتئین غیرطبیعی تولید شده توسط جهش R124C دچار تاخوردگی نادرست و تجمع خودبهخودی میشود و به صورت فیبریلهای آمیلوئیدی نامحلول در لایه بومن و استرومای سطحی رسوب میکند. در مراحل پیشرفته، رسوب به لایههای عمیقتر استروما گسترش مییابد.
رسوب آمیلوئید باعث تغییر در ساختارهای چسبندگی اپیتلیال در قرنیه قدامی میشود و منجر به دژنراسیون سلولهای پایه اپیتلیال و دژنراسیون لایه اپیتلیال همراه با نقص در غشای بومن میگردد. این اختلال ساختاری زمینه پاتولوژیک فرسایش مکرر اپیتلیال قرنیه را تشکیل میدهد.
در ژن TGFBI، تفاوت در محل جهش و اسید آمینه جایگزین شده، تصویر بالینی را تعیین میکند. R124C باعث LCD1، R124H باعث دیستروفی گرانولار قرنیه نوع II (نوع آولینو) و R124L باعث دیستروفی قرنیه Reis-Bücklers میشود5). مکانیسم مولکولی که توسط آن تفاوت تنها یک اسید آمینه، ماده رسوبی (آمیلوئید در مقابل هیالین در مقابل هر دو) و محل رسوب را تعیین میکند، به طور کامل شناخته نشده است، اما تصور میشود که محل جهش در کدام دامنه βig-h3 و تأثیر آن بر پایداری تاخوردگی کلیدی باشد.
در LCD IIIA، جهشهای غالب در لایههای عمیق مانند L527R باعث ایجاد خطوط مشبک ضخیم طنابمانند میشوند و نوع دیررس بدون اختلال اپیتلیال ایجاد میکنند. محلیسازی لایهای رسوبات را میتوان با گرادیان ترشح و انتشار βig-h3 از سلولهای تولیدکننده (سلولهای پایه اپیتلیال) به داخل استروما و تفاوت در پایداری تاخوردگی پروتئین جهشیافته توضیح داد. تصور میشود که R124C مسیر از واسطه تاخوردگی به سمت تشکیل فیبریل آمیلوئید را ترجیح میدهد و آمیلوئید را در اطراف لایه بومن تجمع میدهد5). در مقابل، جهش L527R یک پروتئین نسبتاً پایدار با تاخوردگی نادرست تشکیل میدهد که به آرامی در لایههای عمیقتر استروما رسوب میکند.
به طور سنتی تصور میشد که رسوب آمیلوئید در LCD1 به قرنیه قدامی (لایه بومن تا استرومای سطحی) محدود است. با این حال، بررسیهای پاتولوژیک اخیر نشان داده است که رسوب آمیلوئید در قرنیه خلفی نزدیک غشای دسمه نیز وجود دارد3). رسوب آمیلوئید در قرنیه خلفی ممکن است بر چسبندگی غشای دسمه تأثیر بگذارد و به جداشدگی غشای دسمه در طول جراحی آب مروارید کمک کند3). پیشنهاد شده است که مکانیسم مشابهی که در قرنیه قدامی چسبندگی اپیتلیال را مختل میکند، در بخش خلفی نیز عمل میکند3).
ژلسولین، مولکول عامل سندرم مرتویا (LCD2 سابق)، هم در سیتوپلاسم و هم خارج سلولی وجود دارد و پروتئینی است که از طریق اتصال به اکتین در حرکت سلولی، تقسیم سلولی و آپوپتوز نقش دارد. جهش کلاسیک D187N که نوع فنلاندی نامیده میشود، فنوتیپی با رسوبات شبکهای قرنیه و نوروپاتی کرانیال ایجاد میکند11). جهش جدید p.Glu580Lys که در یک خانواده اسلوونیایی گزارش شده است، در مرز دامنه G4-G5 قرار دارد و با جایگزینی گلوتامات با بار منفی به لیزین با بار مثبت، دافعه الکترواستاتیکی ایجاد کرده و اتصال و پایداری بین دامنهها را کاهش میدهد2). ژلسولین جهشیافته در پلاسما تحت برش غیرطبیعی توسط فورین و MT1-MMP قرار گرفته و قطعات پیشساز آمیلوئید 8 کیلودالتونی و 5 کیلودالتونی آزاد میکند. این قطعات در استرومای قرنیه، پوست، دیواره عروق، اعصاب محیطی و گلومرول کلیه رسوب کرده و علائم چندعضوی مشخصه سندرم مرتویا را ایجاد میکنند2,11). رسوبات قرنیه اغلب قبل از سایر علائم سیستمیک ظاهر میشوند و میتوانند فرصتی برای تشخیص اولیه این بیماری توسط چشمپزشک باشند.
بروز LCD ناشی از جهشهای de novo در ژن TGFBI گزارش شده است1). حتی در موارد بدون سابقه خانوادگی، باید احتمال جهش de novo را در نظر گرفت و تأیید با آزمایش ژنتیک توصیه میشود1). جهش L509P نادر است اما فنوتیپهای متنوعی از شبه دیستروفی قرنیه Reis-Bücklers تا شبه LCD IIIA ایجاد میکند1).
در ژن GSN، علاوه بر جهشهای کلاسیک p.Asp187Asn/Tyr، جهش جدید p.Glu580Lys گزارش شده است که باعث دیستروفی شبکهای قرنیه، شلی پوست، آریتمی قلبی، نفروپاتی و نوروپاتی بینایی همراه با آمیلوئیدوز سیستمیک میشود2).
رسوبات آمیلوئید در قرنیه خلفی بیماران LCD1 وجود دارد و از نظر پاتولوژیک نشان داده شده است که ممکن است بر چسبندگی غشای دسمه تأثیر بگذارد3). در جراحیهای داخل چشمی مانند جراحی آب مروارید، باید به خطر جداشدن غشای دسمه توجه شود.
این یافته پیامدهای بالینی برای ارزیابی اندیکاسیون جراحی آب مروارید و برنامهریزی روش جراحی در بیماران LCD1 دارد.
تکنیکهای جراحی دقیقتری مانند کراتکتومی لایهای با کمک لیزر فمتوثانیه (FLK) و کراتوپلاستی لایهای با کمک لیزر فمتوثانیه (FALK) در حال توسعه هستند 12). این روشها با بهبود صافی سطح برش و کنترل عمق با تکرارپذیری بالا، به عنوان گزینههای مکمل برای PTK سنتی در نظر گرفته میشوند.
از آنجایی که جهش TGFBI یک جهش غالب اتوزومی با عملکرد افزایشی است، siRNA اختصاصی آلل جهشیافته، الیگونوکلئوتیدهای آنتیسنس، و ناکاوت اختصاصی آلل با CRISPR-Cas9 در مرحله مطالعات پیشبالینی بررسی میشوند. قرنیه به دلیل امکان تجویز موضعی و داشتن امتیاز ایمنی، اندام هدف مناسبی برای ژن درمانی است. با این حال، در حال حاضر هیچ کدام به کار بالینی نرسیدهاند و همگی نیاز به تأیید ایمنی و کارایی طولانیمدت در آینده دارند.
ترکیبات مولکولی کوچک هدفگیرنده فرآیند تجمع TGFBIp یا ژلسولین جهشیافته، شپرونهای مولکولی (مانند القاکنندههای Hsp70)، و مهارکنندههای اتصال فیبریل آمیلوئید در مرحله تحقیقات پایه بررسی میشوند. برای آمیلوئیدوز سیستمیک از نوع ژلسولین، داروهایی که مرحله برش ژلسولین جهشیافته در پلاسما را مهار میکنند، در برخی مطالعات پیشبالینی ارزیابی شدهاند 2). انتظار میرود در آینده، چنین درمانهای مولکولی هدفمند به عنوان درمان ریشهای جایگزین روشهای سنتی برداشت فیزیکی (PTK و پیوند قرنیه) شوند.
تجزیه و تحلیل پروتئوم قرنیه با استفاده از طیفسنجی جرمی نشان داده است که در رسوبات LCD1، نه تنها TGFBIp بلکه چندین پروتئین غیرطبیعی دیگر نیز ممکن است همرسوب کنند. برای کاربرد بالینی آینده، نقش بیماریزایی این پروتئینهای همرسوب در حال بررسی است.
Ji YW, Ahn H, Shin KJ, Kim TI, Seo KY, Stulting RD, et al. De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. Journal of clinical medicine. 2022;11(11). doi:10.3390/jcm11113055. PMID:35683443; PMCID:PMC9181583.
Potrc M, Volk M, de Rosa M, Pizem J, Teran N, Jaklic H, Maver A, Drnovsek-Olup B, Bollati M, Vogelnik K, Hocevar A, Gornik A, Pfeifer V, Peterlin B, Hawlina M, Fakin A. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. Int J Mol Sci. 2021;22(3):1084.
Matsumoto A, Fukuoka H, Sotozono C. Descemet’s Membrane Detachment During Cataract Surgery in Lattice Corneal Dystrophy Type I: Histopathological Analysis of Posterior Corneal Involvement. Cureus. 2025;17(3):e81431. doi:10.7759/cureus.81431. PMID:40296953; PMCID:PMC12037203.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
Lisch W, Seitz B. The clinical landmarks of corneal dystrophies. Developments in ophthalmology. 2011;48:9-23. doi:10.1159/000324075. PMID:21540629.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern. San Francisco, CA: American Academy of Ophthalmology; 2024.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea. 2003;22(1):19-21.
Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969;1(4):314-324. PMID:4313418.
Kivelä T, Tarkkanen A, Frangione B, Ghiso J, Haltia M. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja’s syndrome. Invest Ophthalmol Vis Sci. 1994;35(10):3759-3769. PMID:8088963.
Steger B, Romano V, Biddolph S, Willoughby CE, Batterbury M, Kaye SB. Femtosecond laser-assisted lamellar keratectomy for corneal opacities secondary to anterior corneal dystrophies: an interventional case series. Cornea. 2016;35(1):6-13. PMID:26509759. doi:10.1097/ICO.0000000000000665.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.