LCD1(古典型)
初發部位:雙眼瞳孔區的Bowman層至實質淺層出現微細點狀、線狀混濁。
格子狀線:具有雙重輪廓的絲狀、線狀混濁相互纏繞,形成網狀、星狀混濁。
進展期:中央部角膜出現卵黃形或圓形乳白色混濁。
反歸照明法:直接照明下難以看清的半透明細格子狀線清晰浮現。
螢光素染色:上皮黏附性降低導致表面粗糙。
復發性上皮糜爛:由於沉積物累及上皮基底細胞和Bowman膜,因此頻繁發生。

格子狀角膜營養不良(lattice corneal dystrophy, LCD)是一種遺傳性角膜營養不良,澱粉樣物質沉積於角膜基質,產生格子狀線狀混濁。該病歷史悠久,早在1890年代就有記載,在IC3D(國際角膜營養不良分類委員會)第2版臨床遺傳學分類中,統一為LCD1及其變異型(原3型、3A型、1/3A型和4型)4)。
LCD1、顆粒狀角膜營養不良、Reis-Bücklers角膜營養不良和Thiel-Behnke角膜營養不良構成一組疾病,稱為「TGFBI相關營養不良」。致病基因TGFBI(轉化生長因子β誘導基因)位於第5號染色體長臂(5q31),呈體染色體顯性遺傳。TGFBI蛋白(TGFBIp、kerato-epithelin、βig-h3)由角膜上皮細胞產生,分布於角膜全層。在角膜基質中,它參與膠原纖維的組裝。即使同一基因的突變,由於突變位點和胺基酸置換的不同,沉積物(透明蛋白或澱粉樣蛋白)和臨床表現也有顯著差異5)。
LCD1的代表性突變是TGFBI基因第124位精氨酸被半胱氨酸取代的R124C。在變異型LCD IIIA中,已報告有L527R等突變。
角膜中積累的異常蛋白經剛果紅染色呈紅色,在偏光顯微鏡下呈現特有的蘋果綠雙折射,從而確認為澱粉樣蛋白。這一發現自19世紀以來一直是澱粉樣變性的經典組織診斷指標6)。
傳統上稱為「格子狀角膜營養不良2型」的類型是全身性凝溶膠蛋白型澱粉樣變性(GSN-AMYL,Meretoja症候群)的眼部表現,在當前的IC3D分類中被歸類為「家族性澱粉樣變性」,與經典LCD分開處理4,10)。該症候群於1969年由芬蘭的Meretoja首次描述,是一種遺傳性疾病,除角膜格子狀混濁外,還伴有進行性腦神經病變、皮膚鬆弛和全身症狀10,11)。由於臨床上兩者的鑑別很重要,本文一併描述。
| 病型 | 基因 | 代表性突變 | 發病年齡 | 主要表現 |
|---|---|---|---|---|
| LCD1(經典型) | TGFBI(5q31) | R124C | 10~20歲 | 瞳孔區雙重輪廓絲狀混濁,復發性上皮糜爛 |
| LCD IIIA(變異型) | TGFBI(5q31) | L527R 等 | 40歲以後 | 實質深層粗繩狀格子線,無上皮病變 |
| GSN型(Meretoja) | GSN(9q34) | D187N、p.Glu580Lys2) | 30~40歲 | 周邊部放射狀格子線,全身性類澱粉沉積症 |
在日本,頻率高的TGFBI相關角膜營養不良絕大多數是顆粒狀II型(Avellino型、R124H),LCD1與之相比較少。但由於兩者在相同的TGFBI基因上僅相差幾個鹼基而導致病型不同,因此在臨床表現重疊的病例中,建議透過基因檢測確診。日本LCD整體的準確盛行率尚未報告,但在角膜營養不良中屬於相對罕見的類型。
LCD1是由TGFBI基因突變引起的侷限於角膜的類澱粉沉積,1020歲從瞳孔區發病,常伴有復發性上皮糜爛。而Meretoja症候群(舊稱LCD2、GSN型)是由GSN(凝溶膠蛋白)基因突變引起的全身性類澱粉沉積症的眼部表現,3040歲從周邊角膜發病,中央透明性長期保持。Meretoja症候群伴有皮膚鬆弛、面具樣面容、周邊神經病變、心律不整等全身症狀2,10)。在IC3D第2版中,Meretoja症候群被獨立分類,不屬於格子狀角膜營養不良4)。
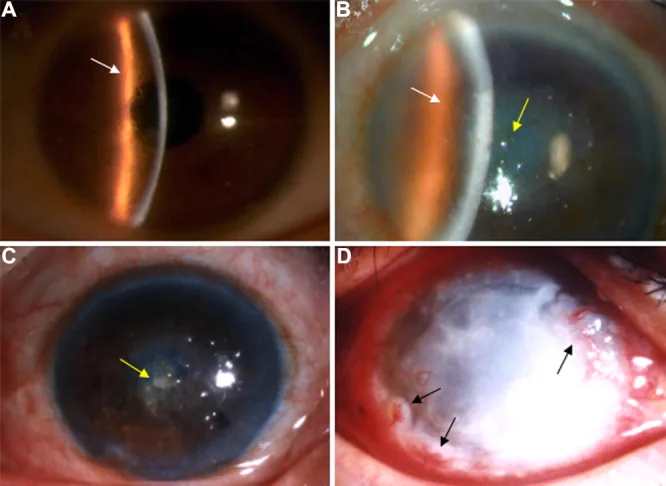
在LCD1中,兒童期多無症狀,僅在裂隙燈顯微鏡的徹照法下可檢測到細微混濁。10~20歲以後反覆出現復發性角膜上皮糜爛(RCE),表現為晨起時的劇烈眼痛、畏光、流淚和異物感。30歲左右角膜中央部實質淺層出現白色混濁,40歲以後視力下降逐漸進展。
在LCD IIIA(變異型)中,通常不發生上皮損傷,主要症狀為40歲以後緩慢的視力下降。
在舊LCD2(Meretoja症候群)中,眼部症狀出現在30~40歲,但嚴重的視力障礙常延遲至60歲以後11)。眼瞼皮膚鬆弛、面具樣面容、進行性腦神經障礙、心臟心律不整等全身症狀常先於或伴隨眼部症狀出現2,10)。
各病型的裂隙燈顯微鏡所見如下所示。
LCD1(古典型)
初發部位:雙眼瞳孔區的Bowman層至實質淺層出現微細點狀、線狀混濁。
格子狀線:具有雙重輪廓的絲狀、線狀混濁相互纏繞,形成網狀、星狀混濁。
進展期:中央部角膜出現卵黃形或圓形乳白色混濁。
反歸照明法:直接照明下難以看清的半透明細格子狀線清晰浮現。
螢光素染色:上皮黏附性降低導致表面粗糙。
復發性上皮糜爛:由於沉積物累及上皮基底細胞和Bowman膜,因此頻繁發生。
LCD IIIA(變異型)
格子狀線:在實質中層至深層出現粗而長的格子狀線,有時呈樹枝狀分支。直接照明下也可觀察到。
表現型:有三種模式:①僅有格子狀線,②僅有小顆粒狀沉積,③兩者混合。同一個體左右眼可呈現不同表現型,也有單眼病例。
上皮:通常不發生上皮損傷。
同合子:L527R同合子的格子狀線更粗,中央顆粒狀沉積更大,但不如R124H(顆粒狀II型)異合與同合之間的差異顯著。
GSN型(Meretoja)
格子狀線:少數缺乏精細感的格子狀沉積從周邊部放射狀出現。
中央透明性:發病後長期保持中央部透明。
上皮糜爛:罕見。
全身表現:出現面具樣面容、伴有運動障礙的突出嘴唇、下垂的耳朵、眼瞼皮膚鬆弛症等臉部變化2)。
在LCD1中,有些病例中央部圓形混濁特別嚴重。有報導稱一名56歲的R124C異合子因中央圓形混濁需要角膜移植。
兒童期LCD1多無症狀,直接照明下難以發現異常。使用裂隙燈顯微鏡的徹照法或反歸照明法進行詳細觀察,可確認中央實質淺層的細微點狀至線狀混濁。對於反覆發生角膜上皮糜爛的兒童,應考慮到LCD1,建議進行包括家族史詢問和父母角膜檢查在內的評估。TGFBI基因檢測有助於確診。
格子狀角膜營養不良的致病基因和代表性突變總結如下。
TGFBI相關(LCD1、LCD IIIA、LCD IV)
基因座:5q31(TGFBI 基因)。
遺傳方式:體染色體顯性遺傳。
LCD1 常見突變:R124C(Arg124Cys)最常見5)。
LCD IIIA 常見突變:已報告 L527R(Leu527Arg)等。存在同合子病例。
新發突變:已報告 TGFBI L509P 的新發突變呈現 LCD IIIA 表現型的病例1)。父母無突變,一個孩子遺傳了該突變1)。
TGFBIp 的角色:由角膜上皮產生,分布於角膜全層,在基質中參與膠原纖維構建5)。
GSN相關(Meretoja症候群,舊稱LCD2)
基因座:9q34(GSN 基因,凝溶膠蛋白)。
遺傳方式:體染色體顯性遺傳。
經典突變:D187N(芬蘭型)最常見,也有報告 p.Asp187Tyr10,11)。
新穎突變:斯洛維尼亞家族報告的 p.Glu580Lys 位於 G4-G5 結構域邊界,因負電荷轉為正電荷導致靜電排斥2)。
臨床表現:除角膜格子狀混濁外,還伴有皮膚鬆弛、心律不整、腎損害和視神經病變的全身性類澱粉沉積症2)。
由於是遺傳性疾病,家族史是最重要的風險因子。但 TGFBI 可能發生新發突變,因此無家族史不能排除1)。遺傳方式為體染色體顯性遺傳,若父母一方帶有突變,子女有 50% 的機率遺傳。無性別差異,LCD1 種族差異不明顯,但 Meretoja 症候群已知在芬蘭有聚集性家族11)。
環境因子的貢獻尚不明確,本病的發生與進展主要由基因型決定。但復發性上皮糜爛的頻率可能因乾燥環境、隱形眼鏡佩戴或外傷而加重。屈光手術(LASIK、SMILE 等)可能導致 TGFBI 相關營養不良快速惡化,術前篩查中有家族史的病例需注意5)。
鑑別LCD1、變異型和GSN型需要綜合裂隙燈所見、組織所見和基因所見。
臨床檢查
裂隙燈顯微鏡:直接照明下早期格子狀線容易漏診。使用徹照法以瞳孔為背景檢測微細混濁,使用反歸照明法檢測半透明的細格子狀線。
螢光素染色:LCD1中由於上皮黏附性降低,染色變得粗糙。也可用於評估上皮糜爛的範圍。
眼前段光學相干斷層掃描(前段OCT):可定量評估沉積物的分層深度。FD-OCT測量的病變深度有助於確定PTK的切除深度1)。
角膜共聚焦顯微鏡:可在細胞水平觀察基質內的沉積物。
確診
基因檢測:檢測TGFBI基因和GSN基因的突變可確定病型。即使表型相同,突變不同也會改變復發和進展的速度,因此直接關係到治療計劃。
病理檢查:剛果紅染色呈紅色,偏光顯微鏡下呈蘋果綠雙折射,可確認為澱粉樣蛋白6)。
免疫組織化學:使用抗TGFBIp抗體和抗凝溶膠蛋白抗體可進行病型鑑別。
家族史詢問:由於是體染色體顯性遺傳,確認父母和同胞的角膜所見有助於診斷。
即使格子狀角膜營養不良表現型相似,但致病基因和突變位點不同,其進展速度、復發頻率、治療選擇和全身併發症的有無會有很大差異。TGFBI突變的LCD1和GSN突變的Meretoja症候群在治療策略和全身檢查的必要性上根本不同2,10)。此外,已有新生突變的報導,僅憑家族史無法確定疾病類型1),因此基因檢測對於確診和分型至關重要。
格子狀角膜營養不良的治療基於IC3D分類的以下階梯式方法。
在兒童期至青年期,無症狀或僅有細微混濁時進行追蹤觀察。每半年至一年透過裂隙燈檢查評估進展。
對於LCD1的核心症狀——復發性上皮糜爛,以下保守治療為首選。
在類澱粉沉積主要位於角膜表層的LCD1中,對於中央混濁嚴重或反覆出現角膜上皮糜爛的病例,使用準分子雷射的光治療性角膜切除術(PTK)是首選7,8)。通常早期不會復發,但隨著時間推移復發不可避免;同一隻眼可進行最多約兩次PTK治療。
在異合子中,復發緩慢,需要再次治療的病例很少。同合子比異合子更容易早期復發。與其他TGFBI相關營養不良一樣,PTK後的復發率隨時間增加,長期觀察中許多病例會確認某種復發表現8)。
作為顯示PTK有效性的病例,在由TGFBI L509P新生突變引起的LCD IIIA病例中,在FD-OCT引導下進行了60 µm的PTK,最佳矯正視力(BCVA)從20/400改善到20/501)。術後45個月時未觀察到視力下降或顯著復發1)。
AAO的角膜水腫和混濁優選實踐模式指出,PTK對於顆粒狀和格子狀角膜營養不良是「合理的選擇」,可能延遲向DALK或全層角膜移植的過渡,但存在術後混濁的風險。重複進行時,考慮聯合使用絲裂黴素C作為抑制復發性疤痕和實質沉積的手段,並警告當消融超過實質前三分之一或剩餘床厚度小於250 µm時,角膜擴張的風險增加7)。
對於反覆復發的病例,或混濁波及實質中深層時,選擇角膜移植。在LCD1中,通常直到40歲以後才適合角膜移植。在LCD中,角膜內皮細胞通常正常,因此根據混濁深度選擇術式。
| 術式 | 適應症 | 特點 |
|---|---|---|
| 表層角膜移植 | 淺層混濁 | 微創,排斥反應少 |
| 深層板層角膜移植(DALK) | 中深層混濁 | 保留內皮,排斥風險低 |
| 穿透性角膜移植(PK) | 全層混濁 | 視力恢復好,但有排斥和復發風險 |
近年來,由於降低排斥反應風險且視力結果與全層角膜移植相當,DALK 已廣泛用作新的首選治療方法。
角膜移植後 LCD 復發是不可避免的,全層角膜移植後的復發率報告為 5 年 17.8%、8 年 26%、15 年 56% 9)。復發性混濁通常侷限於表層,因此可透過 PTK 去除,從而延長再次移植的間隔時間。對於 LCD IIIA(變異型),除非視力受到明顯影響,否則通常不需要治療。
PTK 可以有效去除表層的類澱粉沉積,改善視力並減少復發性上皮糜爛。在一例 LCD IIIA 病例中,60 µm PTK 後最佳矯正視力從 20/400 提高到 20/50,且 45 個月內無復發 1)。雜合子復發緩慢,但純合子早期復發。深層病變無法透過 PTK 去除,因此深層混濁需要 DALK 或全層角膜移植 7)。
LCD1 的核心病理是 TGFBIp(kerato-epithelin、βig-h3)的異常累積。TGFBIp 通常由角膜上皮產生,分佈於角膜全層,在基質中參與膠原纖維構建和細胞黏附的結構蛋白5)。R124C 突變產生的異常蛋白發生錯誤摺疊和自身聚集,以不溶性澱粉樣原纖維的形式沉積在 Bowman 層和基質淺層。進展期時,沉積物向基質深層擴散。
澱粉樣沉積導致前部角膜的上皮黏附結構發生變化,引起上皮基底細胞變性,以及伴有 Bowman 膜缺損的上皮層變性。這種結構破壞是復發性角膜上皮糜爛的病理基礎。
TGFBI 基因中,突變位點和取代胺基酸的差異決定臨床表型。R124C 導致 LCD1,R124H 導致顆粒狀角膜營養不良 II 型(Avellino 型),R124L 導致 Reis-Bücklers 角膜營養不良5)。僅一個胺基酸的差異如何決定沉積物(澱粉樣蛋白 vs 透明蛋白 vs 兩者)和沉積部位的分子機制尚未完全闡明,但突變位點屬於 βig-h3 的哪個結構域以及對摺疊穩定性的影響被認為是關鍵因素。
在 LCD IIIA 中,L527R 等深層優勢突變產生粗繩狀格子線,形成不伴上皮損傷的晚發型。沉積物的層別定位可透過 βig-h3 的產生細胞(上皮基底細胞)向基質內的分泌和擴散梯度,以及突變蛋白摺疊穩定性的差異來解釋。R124C 被認為優先從摺疊中間體走向澱粉樣原纖維形成途徑,在 Bowman 層周圍累積澱粉樣蛋白5)。而 L527R 突變形成相對穩定的錯誤摺疊蛋白,緩慢沉積於更深的基質層。
傳統認為 LCD1 的澱粉樣沉積局限於前部角膜(Bowman 層至基質淺層)。但近年病理學研究顯示,後部角膜近 Descemet 膜處也存在澱粉樣沉積3)。後部角膜的澱粉樣沉積可能影響 Descemet 膜的黏附,並在白內障手術中導致 Descemet 膜剝離3)。提示前部角膜中澱粉樣沉積損害上皮黏附的類似機制也可能在後部起作用3)。
凝溶膠蛋白(gelsolin)是舊LCD2(Meretoja症候群)的致病分子,存在於細胞質和細胞外,是一種透過肌動蛋白結合參與細胞運動、細胞分裂和細胞凋亡的蛋白質。經典的D187N突變被稱為芬蘭型,主要表現為角膜格子狀沉積和腦神經病變11)。在斯洛維尼亞家族中報導的新型p.Glu580Lys突變位於G4-G5結構域邊界,帶負電的麩胺酸被帶正電的離胺酸取代,產生靜電排斥,從而降低結構域間的連接性和穩定性2)。突變型凝溶膠蛋白在血漿中被弗林蛋白酶和MT1-MMP異常切割,釋放出8 kDa和5 kDa的類澱粉前驅片段。這些片段沉積在角膜基質、皮膚、血管壁、周邊神經和腎絲球體中,引起Meretoja症候群特徵性的多器官症狀2,11)。角膜沉積通常先於其他全身症狀,眼科醫師可能是最早診斷該病的醫師。
已有報導TGFBI基因的從頭突變導致LCD的發生1)。即使沒有家族史的病例,也應考慮從頭突變的可能性,建議透過基因檢測確認1)。L509P突變罕見,但呈現從Reis-Bücklers角膜營養不良樣到LCD IIIA樣的多種表現型1)。
在GSN基因中,除了傳統的p.Asp187Asn/Tyr突變外,還報導了新型p.Glu580Lys突變,該突變可導致伴有角膜格子狀營養不良、皮膚鬆弛、心律不整、腎損傷和視神經病變的全身性類澱粉沉積症2)。
病理學研究表明,LCD1患者的後部角膜存在類澱粉沉積,可能影響Descemet膜的黏附3)。在白內障手術等內眼手術時,需注意Descemet膜剝離的風險。
這一發現對LCD1患者白內障手術適應症評估和手術方案制定具有臨床意義。
飛秒雷射輔助板層角膜切除術(FLK)和飛秒雷射輔助板層角膜移植術(FALK)等更精確的手術技術正在開發中12)。這些技術透過提高切除面的平滑度和高度可重複的深度控制,正被定位為傳統PTK的補充選擇。
由於TGFBI突變是體染色體顯性功能獲得型突變,突變等位基因特異性siRNA、反義寡核苷酸以及CRISPR-Cas9介導的等位基因特異性剔除正在臨床前研究階段進行探討。角膜因可局部給藥且具有免疫特權,作為基因治療的靶器官具有優勢。但目前尚無臨床應用的案例,均需要未來對長期安全性和有效性進行驗證。
針對TGFBIp和突變凝溶膠蛋白聚集過程的小分子化合物、分子伴護蛋白(如Hsp70誘導劑)以及類澱粉原纖維結合抑制劑正在基礎研究階段進行探討。對於系統性凝溶膠蛋白型類澱粉沉積症,一些抑制血漿中突變凝溶膠蛋白裂解步驟的藥物正在部分臨床前試驗中進行評估2)。未來,這類分子標靶治療有望成為取代傳統物理切除(PTK和角膜移植)的根本性治療。
利用質譜法進行的角膜蛋白質體學分析表明,在LCD1的沉積物中,不僅TGFBIp,多種異常蛋白也可能共同沉積。為了未來的臨床應用,正在闡明這些共沉積蛋白的病理貢獻。