患病率的地區差異
美國:罕見,約每25萬人0.3例 2,3)
冰島:約每25萬人19例,是全球最高頻率的地區之一 5,6)
高患病率地區:南印度、沙烏地阿拉伯、冰島和北歐頻率較高 5,7)
其他地區:相對罕見。近親結婚或複合雜合子發病

斑狀角膜失養症(macular corneal dystrophy:MCD)是一種遺傳性角膜失養症,其特徵是糖胺聚醣(主要是硫酸角質素)在角膜基質中積聚。它呈體染色體隱性遺傳,由位於第16號染色體長臂(16q22)的CHST6基因突變引起1,3)。曾稱為Groenouw角膜失養症II型或Fehr角膜失養症。
與許多其他角膜基質失養症(顆粒狀、格子狀)為體染色體顯性遺傳不同,本病的特徵是體染色體隱性遺傳。在日本,它與顆粒狀角膜失養症(I型、II型)、格子狀角膜失養症(I型、IIIA型)和膠滴狀角膜失養症並稱為四大角膜失養症,約佔所有角膜失養症的96%。其中前兩者為體染色體顯性遺傳,後兩者(膠滴狀和斑狀)為體染色體隱性遺傳。
在IC3D(國際角膜營養不良分類委員會)分類中,MCD被定位為一種基質營養不良 1)。Aggarwal等人在2018年《Survey of Ophthalmology》的綜述中,將本病詳述為「罕見但嚴重影響視功能的基質營養不良」,並總結了診斷和治療體系 4)。在IC3D分類第二版中,角膜營養不良分為1至4類,根據致病基因、病理學發現和臨床證據強度進行區分 1)。MCD因CHST6基因突變的鑑定被歸類為第1類(基因層次已確立的營養不良)。
本病的歷史背景方面,1890年由Groenouw首次描述,後來出現了將顆粒狀營養不良稱為「I型」、斑狀營養不良稱為「II型」的慣例。1938年Jones和Zimmerman將其確立為獨立疾病,2000年Akama等人鑑定出CHST6基因,闡明了其分子基礎 3)。
全球範圍內地區差異很大,患病率高的地區有家族聚集性。這是一種相對罕見的疾病。
患病率的地區差異
美國:罕見,約每25萬人0.3例 2,3)
冰島:約每25萬人19例,是全球最高頻率的地區之一 5,6)
高患病率地區:南印度、沙烏地阿拉伯、冰島和北歐頻率較高 5,7)
其他地區:相對罕見。近親結婚或複合雜合子發病
免疫表現型
MCD的免疫表現型通過使用抗硫酸角質素單株抗體檢測角膜和血清中的硫酸角質素量進行分類 2,3)。
| 表現型 | 角膜硫酸角質素 | 血清硫酸角質素 |
|---|---|---|
| I型 | 陰性 | 陰性 |
| IA型 | 陽性(細胞內) | 陰性 |
| II型 | 陽性 | 陽性 |
大多數患者屬於I型或IA型。然而,臨床上區分這些亞型並不重要,且無法透過檢查結果鑑別2,8)。
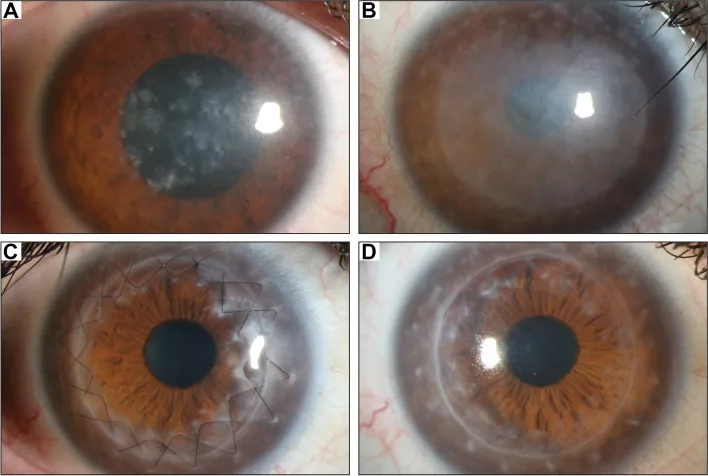
臨床所見的典型進展模式如下。
臨床上,角膜實質可見瀰漫性細小沉積,呈磨玻璃樣混濁。隨著進展,混濁累及實質全層,並從中央向周邊擴散。之後,除了淡薄混濁外,實質淺層至深層可見大量灰白色、形狀不規則的小混濁。9)
初期所見
斑點狀混濁:角膜中央部實質淺層出現灰白色小斑點狀混濁
磨玻璃樣混濁:角膜實質可見瀰漫性淡薄混濁
邊界不清:混濁邊緣不清晰,與正常實質的邊界模糊
進展期表現
全層侵犯:混濁遍及整個基質層
向周邊擴展:混濁從中央向周邊擴散
角膜變薄:中央角膜厚度減少
內皮及Descemet膜沉積:深層結構也有異常物質積聚
裂隙燈顯微鏡下,整個角膜呈瀰漫性混濁,其中可見灰白色不規則斑片狀沉積物。以裂隙光做光學切面時,可見中央部沉積物位於淺層,周邊部位於深層的特徵性分布。斑片狀病變多呈同心圓狀出現8)。
混濁可累及角膜緣,這是與其他角膜營養不良的重要鑑別點。顆粒狀角膜營養不良和格子狀角膜營養不良的角膜緣常保持透明,而MCD的整個角膜包括角膜緣常發生混濁2,8)。不規則散光與前基質沉積有關,也可出現角膜知覺減退。由於內皮也有異常物質沉積,進展期病例可因內皮功能下降而併發基質水腫8)。
自然病程因人而異,但常經歷以下階段:
MCD是一種進行性疾病,一生中視功能持續下降,這與視力損害較輕的顆粒狀角膜營養不良I型和部分格子狀角膜營養不良亞型的預後顯著不同4,8)。
致病基因是 CHST6(碳水化合物磺基轉移酶6)3)。位於16q22,編碼一種將硫酸基轉移到角膜蛋白聚醣上N-乙醯葡萄糖胺的酶。該基因的突變類型非常多,包括錯義突變、無義突變、框架位移突變和5’上游區域缺失等,已在不同民族中報告3,7)。
由於是體染色體隱性遺傳,通常先證者的父母雙方都是攜帶者。在近親結婚較多的地區或群體中,發生率往往較高。不同家族間通婚產生的複合雜合子也可能發病7)。
Akama等人於2000年將CHST6鑑定為本病的致病基因,並表明免疫表現型I型和II型均由同一基因座的突變引起3)。這一發現表明免疫表現型的差異是由單一基因的不同突變模式決定的,是一項重要發現,為後續的基因診斷體系奠定了基礎。
已報告的突變類型超過200種,其中錯義突變最多。Sultana等人在南印度患者群中鑑定出大量新突變,表明該地區的高發生率是由於區域特異性突變的積累所致7)。沙烏地阿拉伯和冰島也報告了類似的區域性聚集,認為人群的歷史背景(創始者效應和近親結婚習俗)對發生率有貢獻5,6,7)。
發生率因地區而異,是一種相對罕見的疾病9)。與顆粒狀角膜營養不良等其他角膜營養不良相比,病例較少,且往往在具有複合雜合子或近親結婚背景的家庭中報告。
懷疑MCD的線索是「雙眼性、進行性、整個角膜瀰漫性混濁的青春期至年輕成人」。首先詳細詢問自覺症狀(視力下降、畏光、刺激感)的經過、家族史、近親結婚史。然後進行裂隙燈顯微鏡下的角膜所見評估、內皮功能評估,必要時進行基因檢測,這是標準流程。
這是診斷的基礎檢查。如果看到無充血或角膜水腫的雙眼角膜混濁,應懷疑角膜營養不良。MCD具有以下特徵性所見:
許多角膜營養不良在裂隙燈水平表現為不連續的病變(沉積物之間有透明部分),而MCD則例外地呈現瀰漫性混濁模式。它與格子狀角膜營養不良I型和膠樣滴狀角膜營養不良一起,被特徵化為「瀰漫性觀察到的角膜沉積」的代表性例子。
CHST6基因分析有助於確診。可在經認證的醫療機構進行。
組織學上,阿爾新藍和膠體鐵染色呈陽性,角膜基質內角膜細胞內外可見瀰漫性低硫酸化糖胺聚糖積聚2,8)。可見Bowman層斷裂,進展期病例內皮細胞內也可見異常物質。Descemet膜上也可出現滴狀角膜(guttae)樣表現。
| 疾病 | 遺傳方式 | 特徵 |
|---|---|---|
| 顆粒狀角膜營養不良 | 體染色體顯性 | 邊界清晰的顆粒狀沉積物(之間有透明區域) |
| 格子狀角膜營養不良I型 | 體染色體顯性 | 線狀和網狀格子線(類澱粉蛋白) |
| 膠樣滴狀角膜失養症 | 體染色體隱性遺傳 | 桑椹樣或帶狀隆起性病變 |
| Schnyder結晶狀角膜失養症 | 體染色體顯性遺傳 | 針狀結晶,合併高脂血症 |
| 先天性角膜基質失養症 | 體染色體顯性遺傳 | 出生時即存在,基質增厚 |
| 全身性黏多醣症(角膜型) | 隱性/X染色體連鎖遺傳 | 伴有全身症狀 |
此外,後部不定形角膜失養症(PACD)和Descemet膜前角膜失養症(PDCD)也需鑑別。全身性黏多醣症(如Hurler、Scheie、Morquio症候群等)也可能引起角膜混濁,因此需要結合全身表現進行評估8)。
MCD的治療目標可歸納為三點:(1) 維持和恢復視功能,(2) 通過穩定眼表緩解疼痛和刺激症狀,(3) 預防併發症(上皮糜爛、感染)。由於沒有根本性的病因治療,根據病程和症狀進行階段性干預。
目前尚無抑制本病進展的藥物療法8)。以下措施用於緩解症狀。
對於視力進行性下降的病例,角膜移植術是唯一的根治手段。根據有無內皮病變選擇術式。
在MCD中,內皮細胞也可能有異常沉積,因此當病變累及深層時,傾向於選擇PKP而非DALK7)。伴有內皮異常時,全層角膜移植是適應症。術前詳細的內皮評估(鏡面顯微鏡和共聚焦顯微鏡)是決定術式的關鍵。
DALK後的復發率較低,報告約為5%。PKP後的植片存活率良好,許多報告顯示可長期存活,但也有報告指出術後數年至十數年後異常物質會重新沉積在植片上8)。Al-Swailem等人在沙烏地阿拉伯的系列研究中,MCD患者PKP後的植片存活率良好,但部分病例在長期追蹤中觀察到復發8)。此外,Reinhart等人的AAO綜述顯示,對於實質層營養不良,DALK可達到與PKP相當或更好的視力恢復,並同時降低內皮細胞丟失率9)。Unal等人的多中心研究也報告了DALK對包括MCD在內的實質層營養不良的有效性。
一般來說,角膜植片術後復發是由於原發病的分子病理狀態殘留在宿主側所致。DALK保留了宿主的內皮和Descemet膜前層,因此在內皮受累的病例中,病變可能進展,需注意。術後需要長期定期觀察(視力、裂隙燈檢查、內皮細胞密度測量)。
CHST6基因編碼碳水化合物磺基轉移酶6 3)。該酶負責將硫酸基團轉移到角質素分子上的N-乙醯葡糖胺,是角膜蛋白聚醣中硫酸角質素(KS)正常合成所必需的。
基因突變導致酶活性喪失時,會合成硫酸化不足的低硫酸化角質素硫酸鹽。這種異常的角質素硫酸鹽溶解度低,在角膜基質中的角膜細胞內外異常沉積2,3)。
硫酸角質素的量和質異常會導致以下連鎖病理變化。
糖胺聚醣的積聚在基質角膜細胞的細胞內和細胞外均可觀察到,隨著病變進展,可擴散至Bowman層、Descemet膜和內皮細胞。在I型中,耳廓軟骨中也觀察到酶活性降低,提示可能是全身性硫酸角質素代謝異常的部分表現2)。但臨床上全身症狀少見,通常作為以角膜為主要症狀的局限性疾病處理。
在角膜蛋白聚醣中,lumican負責將膠原纖維直徑控制在約25 nm,而keratocan和mimecan維持纖維間距均勻。當這些SLRP的硫酸化側鏈短而不完整時,膠原纖維粗細不一,間距也不均勻。結果導致角膜基質內光散射增加,臨床表現為磨玻璃樣混濁。
角膜透明性的維持經典地由Maurer格子理論解釋,即膠原纖維整齊的格子狀排列通過光干涉相消實現。在MCD中,這種格子結構因SLRP異常而破壞,導致透明性喪失2,4)。
近年來的基礎研究報告稱,CHST6突變導致的自噬功能障礙可能誘導角膜細胞焦亡(發炎性細胞死亡),促進疾病進展8)。其他角膜營養不良(如顆粒狀II型)中也報導了類似的自噬異常,作為角膜營養不良的共同病理機制受到關注。
基因標靶治療已被提出作為一種永久性治療策略8)。在Meesmann角膜上皮營養不良中,使用CRISPR/Cas9進行基因編輯的基礎研究正在進展中,未來也可能成為MCD的治療選擇。然而,臨床應用仍面臨許多挑戰,包括對正常等位基因的意外編輯(脫靶效應)、建立向角膜基質細胞高效遞送基因的方法,以及長期安全性的驗證。
一種透過酶學方法去除角膜內積聚的低硫酸化硫酸角質素的方法正在基礎研究中。這借鑒了全身性黏多醣症中已應用的酶替代療法概念,並將其局部應用於角膜,但目前尚無臨床應用的報告。
Descemet膜剝離角膜內皮移植術(DSAEK)和Descemet膜角膜內皮移植術(DMEK)主要是針對Fuchs角膜內皮營養不良和大皰性角膜病變發展而來的手術方式,但將其應用於MCD內皮病變先行病例也是未來的研究課題。目前,對於這種同時累及基質和內皮的疾病,穿透性角膜移植術(PKP)是現實的選擇。未來,有學者提出了「序貫角膜移植術」的概念,即分階段聯合使用僅置換基質的深層板層角膜移植術(DALK)和僅置換內皮的DMEK,但這些仍處於研究階段。
據報導,免疫表型II型MCD患者的血清硫酸角質素水平低於正常值,其作為全身代謝標誌物的有用性正在討論中2)。未來有望應用於血液篩查和CHST6突變家系的早期攜帶者診斷。此外,利用人工智慧自動分析裂隙燈影像也是可能有助於此類罕見疾病早期發現的研究領域。
由於該病為體染色體隱性遺傳,患者同胞中約有25%可能發病。利用CHST6基因檢測,可以進行家族內篩查以及關於婚姻和懷孕的遺傳諮詢。對於有近親結婚史或家族內有類似角膜混濁病例的家系,建議早期轉診至專科醫師。