Die makuläre Hornhautdystrophie (macular corneal dystrophy: MCD) ist eine erbliche Hornhautdystrophie, bei der sich Glykosaminoglykane (hauptsächlich Keratansulfat) im Hornhautstroma ansammeln. Sie wird autosomal-rezessiv vererbt und wird durch eine Mutation des CHST6-Gens auf dem langen Arm von Chromosom 16 (16q22) verursacht1,3). Früher wurde sie auch als Groenouw-Hornhautdystrophie Typ II oder Fehr-Hornhautdystrophie bezeichnet.
Im Gegensatz zu vielen anderen Hornhautstromadystrophien (granulär, gittrig), die autosomal-dominant vererbt werden, zeichnet sich diese Erkrankung durch einen autosomal-rezessiven Erbgang aus. In Japan zählt sie zusammen mit der granulären Hornhautdystrophie (Typ I, II), der gittrigen Hornhautdystrophie (Typ I, IIIA) und der gelatinösen tropfenartigen Hornhautdystrophie zu den vier großen Hornhautdystrophien, die etwa 96% aller Hornhautdystrophien ausmachen. Von diesen sind die ersten beiden autosomal-dominant, die letzten beiden (gelatinöse tropfenartige und makuläre) autosomal-rezessiv.
Gemäß der IC3D-Klassifikation (International Committee for Classification of Corneal Dystrophies) wird die MCD als eine Form der stromalen Dystrophie eingeordnet 1). In einem Übersichtsartikel von Aggarwal et al. im Survey of Ophthalmology 2018 wird diese Erkrankung als „seltene, aber die Sehfunktion stark beeinträchtigende stromale Dystrophie“ beschrieben und das diagnostische und therapeutische System zusammengefasst 4). In der zweiten Auflage der IC3D-Klassifikation werden Hornhautdystrophien in die Kategorien 1 bis 4 eingeteilt, basierend auf der Evidenzstärke des verursachenden Gens, der pathologischen Befunde und des klinischen Bildes 1). Die MCD wird aufgrund der Identifizierung von CHST6-Genmutationen als Kategorie 1 (auf genetischer Ebene etablierte Dystrophie) eingestuft.
Historisch gesehen wurde diese Erkrankung erstmals 1890 von Groenouw beschrieben, und später entstand die Praxis, die granuläre Dystrophie als „Typ I“ und die makuläre Dystrophie als „Typ II“ zu bezeichnen. 1938 wurde sie von Jones und Zimmerman als eigenständige Erkrankung etabliert, und im Jahr 2000 identifizierten Akama et al. das CHST6-Gen, wodurch die molekulare Grundlage geklärt wurde 3).
Weltweit gibt es große regionale Unterschiede; in Gebieten mit hoher Prävalenz tritt eine familiäre Häufung auf. Es handelt sich um eine relativ seltene Erkrankung.
Regionale Unterschiede in der Prävalenz
USA : etwa 0,3 Personen pro 250.000 Einwohner, selten 2,3)
Island : etwa 19 Personen pro 250.000 Einwohner, eine der weltweit häufigsten Regionen 5,6)
Hochprävalenzregionen : hohe Häufigkeit in Südindien, Saudi-Arabien, Island und den nordischen Ländern 5,7)
Andere Regionen : relativ selten. Tritt bei Blutsverwandtschaft oder compound Heterozygotie auf
Immunphänotyp
Typ I : Keratansulfat negativ in Hornhaut und Serum 2)
Typ IA : positiv in Hornhaut-Keratinozyten, negativ im Serum 2)
Typ II : Keratansulfat positiv in Hornhaut und Serum 2)
Klinisches Bild : Alle drei Typen haben denselben Phänotyp und sind mit der Spaltlampe nicht unterscheidbar 2,8)
Der Immunphänotyp der MCD wird anhand der Menge an Keratansulfat in Hornhaut und Serum unter Verwendung von monoklonalen Anti-Keratansulfat-Antikörpern klassifiziert 2,3).
Phänotyp
Korneales Keratansulfat
Serum-Keratansulfat
Typ I
Negativ
Negativ
Typ IA
Positiv (intrazellulär)
Negativ
Typ II
Positiv
Positiv
Die meisten Patienten werden als Typ I oder IA klassifiziert. Klinisch ist die Unterscheidung dieser Subtypen jedoch nicht wichtig und sie können durch die Untersuchungsbefunde nicht differenziert werden2,8).
QWas unterscheidet die makuläre Hornhautdystrophie von anderen Hornhautdystrophien?
A
Der größte Unterschied ist der autosomal-rezessive Erbgang. Die granuläre und gittrige Hornhautdystrophie sind autosomal-dominant, aber diese Erkrankung erfordert Mutationen in beiden Allelen des CHST6-Gens. Zudem zeigt sie eine diffuse Mattglas-Trübung, eine Eintrübung der gesamten Hornhaut, und im Spaltlampenlicht sind die Ablagerungen zentral oberflächlich und peripher tief.
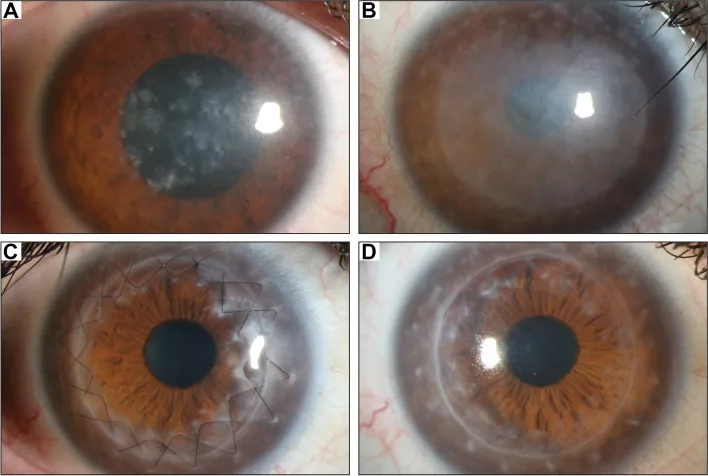
Gassel CJ, et al. Histological findings of corneal tissue after failed phototherapeutic keratectomy in macular corneal dystrophy - a case report. BMC Ophthalmol. 2022. Figure 1. PMCID: PMC9080147. License: CC BY.
Spaltlampenfoto, das eine diffuse grauweiße Trübung und fleckige Ablagerungen von der Mitte bis zur gesamten Hornhaut zeigt. Dies zeigt die typischen klinischen Befunde der makulären Hornhautdystrophie und erleichtert das Verständnis der Hornhauttrübung, die zu Sehverschlechterung führt.
Das typische Fortschreiten der klinischen Befunde ist wie folgt.
Klinisch zeigt sich eine diffuse feine Ablagerung im Hornhautstroma, das wie mattiertes Glas getrübt ist. Die Trübung schreitet fort, um die gesamte Stromadicke zu betreffen, und breitet sich von der Mitte zur Peripherie aus. Danach treten zusätzlich zur leichten Trübung zahlreiche kleine, unregelmäßig geformte grauweiße Trübungen in den oberflächlichen bis tiefen Stromaschichten auf.9)
Frühbefunde
Fleckige Trübungen : kleine grauweiße fleckige Trübungen treten in den oberflächlichen Stromaschichten der Hornhautmitte auf
Mattglasartige Trübung : diffuse leichte Trübung des Hornhautstromas
Unscharfe Grenzen : die Ränder der Trübung sind undeutlich, die Grenze zum normalen Stroma ist unklar
Fortgeschrittene Befunde
Ausdehnung auf die gesamte Dicke: Die Trübung erstreckt sich über die gesamte Stromadicke
Ausbreitung zur Peripherie: Die Trübung breitet sich von der Mitte zur Peripherie aus
Hornhautverdünnung: Die Dicke der zentralen Hornhaut nimmt ab
Ablagerungen auf Endothel und Descemet-Membran: Auch in tiefen Strukturen sammeln sich abnorme Substanzen an
Im Spaltlampenmikroskop erscheint die gesamte Hornhaut diffus getrübt mit unregelmäßigen grau-weißen fleckigen Ablagerungen. Im optischen Schnitt liegen die Ablagerungen zentral oberflächlich und peripher tief, eine charakteristische Verteilung. Die Läsionen treten oft in konzentrischen Mustern auf8).
Die Trübung kann sich bis zum Limbus erstrecken, ein wichtiger Unterschied zu anderen Hornhautdystrophien. Bei der granulären und gittrigen Hornhautdystrophie bleibt der Limbus oft klar, während bei MCD die gesamte Hornhaut bis zum Limbus häufig getrübt ist2,8). Ein unregelmäßiger Astigmatismus ist mit den vorderen Stromaablagerungen verbunden, und eine verminderte Hornhautsensibilität kann auftreten. Endothelablagerungen können in fortgeschrittenen Fällen zu einem Stromaödem durch Endotheldysfunktion führen8).
Der natürliche Verlauf variiert individuell, folgt jedoch oft diesen Phasen.
Frühe Kindheit (asymptomatische Phase): Die Genmutation ist von Geburt an vorhanden, aber Spaltlampenbefunde sind spärlich und der Patient ist asymptomatisch
Schulalter bis Jugend (frühe Trübungsphase): Im vorderen Stroma tritt eine diffuse leichte Trübung auf, später sind fleckige Ablagerungen erkennbar
10–30 Jahre (Sehverschlechterungsphase): Die Trübung schreitet fort, und der Patient bemerkt eine Sehverschlechterung
30–40 Jahre (fortgeschrittene Phase): Die Trübung erstreckt sich über das gesamte Stroma und den Limbus, mit verminderter Hornhautsensibilität, Verdünnung und unregelmäßigem Astigmatismus
Mittleres bis höheres Alter (Phase der Hornhauttransplantation): Die Sehfunktion ist so weit eingeschränkt, dass sie den Alltag beeinträchtigt, und eine Transplantation wird in Betracht gezogen
MCD ist eine fortschreitende Erkrankung mit kontinuierlichem Sehverlust im Laufe des Lebens, was sie von der granulären Typ-I- und gittrigen Hornhautdystrophie (einige Subtypen) unterscheidet, bei denen die Sehbehinderung mild bleibt4,8).
Das verantwortliche Gen ist CHST6 (Carbohydrate Sulfotransferase 6)3). Es liegt auf 16q22 und kodiert ein Enzym, das eine Sulfatgruppe auf N-Acetylglucosamin an Proteoglykanen der Hornhaut überträgt. Die Mutationen dieses Gens sind sehr vielfältig: Missense-, Nonsense-, Frameshift-Mutationen, Deletionen im 5’-Bereich, die in verschiedenen ethnischen Gruppen berichtet wurden3,7).
Die Vererbung ist autosomal-rezessiv, daher sind in der Regel beide Eltern des Probanden Träger. In Regionen oder Gruppen mit häufigen Blutsverwandtenehen ist die Inzidenz besonders hoch. Auch compound-heterozygote Individuen aus Ehen zwischen verschiedenen Familien können erkranken7).
Akama et al. identifizierten CHST6 im Jahr 2000 als ursächliches Gen und zeigten, dass sowohl der Immunphänotyp I als auch II durch Mutationen desselben Genlocus verursacht werden3). Diese Entdeckung deutete darauf hin, dass die Unterschiede im Immunphänotyp durch unterschiedliche Mutationsmuster eines einzelnen Gens bestimmt werden, was die Grundlage für die spätere genetische Diagnostik bildete.
Es wurden über 200 Mutationen berichtet, wobei Missense-Mutationen am häufigsten sind. Sultana et al. identifizierten zahlreiche neue Mutationen bei südindischen Patienten und zeigten, dass die hohe Häufigkeit in dieser Region auf eine Anhäufung populationsspezifischer Mutationen zurückzuführen ist7). Ähnliche regionale Häufungen wurden aus Saudi-Arabien und Island berichtet, was darauf hindeutet, dass der historische Hintergrund der Bevölkerung (Gründereffekt, Blutsverwandtenehen) zur Erkrankungshäufigkeit beiträgt5,6,7).
Die Inzidenz variiert je nach Region; es ist eine relativ seltene Erkrankung9). Im Vergleich zu anderen Hornhautdystrophien wie der granulären Hornhautdystrophie gibt es weniger Fälle, die häufig aus Familien mit compound-Heterozygotie oder Blutsverwandtschaftshintergrund berichtet werden.
Familienanamnese: autosomal-rezessive Vererbung, beide Eltern müssen Träger sein
Blutsverwandtschaft: erhöht die Inzidenz
Geografische Region: hohe Prävalenz in Südindien, Saudi-Arabien, Island, Skandinavien5,7)
QIst ein Gentest notwendig?
A
Ein Test des CHST6-Gens ist zur Bestätigung der Diagnose nützlich. Er kann in akkreditierten medizinischen Einrichtungen durchgeführt werden. In vielen Einrichtungen steht jedoch die klinische Diagnose mittels Spaltlampenmikroskopie im Vordergrund. Der Test ist nützlich für die Risikobewertung bei zukünftigen Kindern oder zur Bestätigung der Diagnose bei atypischen klinischen Befunden.
Der Verdacht auf MCD ergibt sich aus der Trias: beidseitig, fortschreitend, diffuse Trübung der gesamten Hornhaut bei Jugendlichen oder jungen Erwachsenen. Zunächst werden der Verlauf der subjektiven Symptome (Sehverschlechterung, Lichtempfindlichkeit, Reizgefühl), die Familienanamnese und Konsanguinität detailliert erfragt. Anschließend erfolgen die Beurteilung der Hornhaut mittels Spaltlampe, die Bewertung der Endothelfunktion und gegebenenfalls ein Gentest.
Dies ist die grundlegende Untersuchung für die Diagnose. Bei beidseitiger Hornhauttrübung ohne Rötung oder Hornhautödem sollte an eine Hornhautdystrophie gedacht werden. Die MCD zeigt folgende charakteristische Befunde:
Diffuse Mattglastrübung : Die gesamte Hornhaut ist diffus getrübt
Fleckige Ablagerungen : Zahlreiche unregelmäßige grau-weiße Trübungen
Konzentrische Verteilung : Im Spaltlichtschnitt zentral oberflächlich, peripher tief
Limbusinfiltration : Die Trübung kann bis zum Limbus reichen
Endothelanomalien : In fortgeschrittenen Fällen können tropfenförmige Ablagerungen auftreten
Während die meisten Hornhautdystrophien im Spaltlampenbild als diskontinuierliche Läsionen (mit transparenten Bereichen zwischen den Ablagerungen) erscheinen, zeigt die MCD ausnahmsweise ein diffuses Trübungsmuster. Sie wird zusammen mit der gittrigen Hornhautdystrophie Typ I und der gelatinösen tropfenförmigen Hornhautdystrophie als typisches Beispiel für “diffus beobachtete Hornhautablagerungen” charakterisiert.
Spiegelmikroskop : Beurteilung der Endothelzelldichte und -morphologie. Das Ausmaß der Endothelerkrankung beeinflusst direkt die Wahl der Operationstechnik
Hornhaut-Pentacam (Scheimpflug-Kamera) : liefert Dichtekarten der gesamten Hornhautdicke, nützlich für die dreidimensionale Beurteilung von Trübungsbereichen
Histologisch zeigen sich positive Färbungen mit Alcianblau und kolloidalem Eisen, und es wird eine diffuse Ansammlung von hyposulfatierten Glykosaminoglykanen innerhalb und außerhalb der Keratozyten des Hornhautstromas beobachtet2,8). Es können Risse in der Bowman-Membran auftreten, und in fortgeschrittenen Fällen finden sich auch abnorme Substanzen in den Endothelzellen. Auf der Descemet-Membran können auch guttae-ähnliche Befunde auftreten.
Darüber hinaus kommen auch die posteriore polymorphe Hornhautdystrophie (PPCD) und die prä-Descemet-Hornhautdystrophie (PDCD) für die Differentialdiagnose in Frage. Systemische Mukopolysaccharidosen (Hurler-, Scheie-, Morquio-Syndrom usw.) können ebenfalls Hornhauttrübungen verursachen, daher ist eine Bewertung einschließlich systemischer Befunde erforderlich8).
QWie wird die makuläre Hornhautdystrophie diagnostiziert?
A
Die klinische Diagnose erfolgt durch Spaltlampenmikroskopie, die eine diffuse mattglasartige Hornhauttrübung und makuläre Ablagerungen zeigt, sowie durch die bilaterale, progressive Natur, Familienanamnese und das Erkrankungsalter (10–30 Jahre). Zur Bestätigung ist ein Gentest des CHST6-Gens hilfreich.
Die Ziele der Behandlung der MCD sind: (1) Erhalt/Wiederherstellung der Sehfunktion, (2) Linderung von Schmerzen und Reizsymptomen durch Stabilisierung der Augenoberfläche und (3) Vorbeugung von Komplikationen (Epithelerosion, Infektion). Da keine kausale Behandlung existiert, erfolgt eine stufenweise Intervention je nach Stadium und Symptomen.
Es gibt keine etablierte medikamentöse Therapie, um das Fortschreiten dieser Erkrankung zu verlangsamen8). Zur Symptomlinderung werden folgende Maßnahmen durchgeführt:
Künstliche Tränen : Schutz der Augenoberfläche und Vorbeugung von Trockenheit
Bei fortschreitender Sehverschlechterung ist die Hornhauttransplantation die einzige kurative Maßnahme. Die Operationstechnik wird je nach Vorhandensein oder Fehlen einer Endothelbeteiligung ausgewählt.
Tiefe anteriore lamelläre Keratoplastik (DALK) : Mittel der Wahl bei Fällen ohne Endothelbeteiligung8,9). Durch den Erhalt des eigenen Hornhautendothels des Patienten ist das Risiko einer Transplantatabstoßung geringer als bei einer perforierenden Keratoplastik. Auch im Evaluationsbericht der AAO (American Academy of Ophthalmology) wird DALK bei Stromadystrophien als gleichwertig zur PKP hinsichtlich der Sehfunktionswiederherstellung bei geringerem Endothelverlust bewertet8). Die Rezidivrate nach DALK wird als niedrig berichtet, und aufgrund des Austauschs des Hornhautstromas des Empfängers ist ein Rezidiv unwahrscheinlich.
Perforierende Keratoplastik (PKP) : Indiziert bei fortgeschrittenen Fällen mit Ablagerungen abnormaler Substanzen im Endothel oder der Descemet-Membran sowie bei Fällen mit hochgradiger zentraler Hornhautverdünnung7). Das Durchschnittsalter für die erste PKP bei MCD wird mit 30–40 Jahren angegeben7), und die Transplantatüberlebensrate ist gut.
Phototherapeutische Keratektomie (PTK) : Symptomatische Behandlung rezidivierender Hornhautepithel erosionen und oberflächlicher narbiger Trübungen. Es ist jedoch auf die Induktion von Hyperopie und Stromatrübung zu achten.
Da bei MCD auch in Endothelzellen abnormale Substanzen abgelagert werden können, wird bei tiefreichender Erkrankung tendenziell PKP gegenüber DALK bevorzugt7). Bei Endothelanomalien ist eine perforierende Keratoplastik indiziert. Eine detaillierte präoperative Endothelbeurteilung (Spiegelmikroskopie, konfokale Mikroskopie) ist entscheidend für die Wahl der Operationstechnik.
Die Rezidivrate nach DALK ist niedrig, etwa 5 %. Die Transplantatüberlebensrate nach PKP ist gut, mit langfristigem Überleben in vielen Berichten, obwohl auch Fälle von erneuter Ablagerung abnormaler Substanz auf dem Transplantat Jahre bis Jahrzehnte nach der Operation beschrieben wurden 8). In der saudi-arabischen Serie von Al-Swailem et al. war die Überlebensrate nach PKP bei MCD gut, aber in einigen Fällen wurde ein Rezidiv im Langzeitverlauf beobachtet 8). Darüber hinaus zeigte der AAO-Review von Reinhart et al., dass DALK bei Stromadystrophien eine gleichwertige oder bessere Sehfunktion und eine geringere Endothelverlustrate im Vergleich zu PKP bietet 9). Eine multizentrische Studie von Unal et al. berichtete ebenfalls über die Wirksamkeit von DALK bei Stromadystrophien einschließlich MCD.
Im Allgemeinen tritt ein postoperatives Rezidiv von Hornhauttransplantaten auf, weil die molekulare Pathogenese der Grunderkrankung im Wirt verbleibt. Bei DALK werden das Endothel und die prädescemetale Schicht des Wirts erhalten, daher ist zu beachten, dass die Erkrankung bei Fällen mit Endothelbeteiligung fortschreiten kann. Postoperativ ist eine langfristige regelmäßige Nachsorge (Sehschärfe, Spaltlampe, Endothelzellzählung) erforderlich.
QSollte man DALK oder PKP wählen?
A
Wenn das Endothel oder die Descemet-Membran nicht betroffen ist, ist die tiefe anteriore lamelläre Keratoplastik (DALK) die erste Wahl. DALK erhält das eigene Hornhautendothel, wodurch das Abstoßungsrisiko geringer ist, und die postoperative Rezidivrate wird mit etwa 5 % angegeben. Bei Fällen, bei denen auch das Endothel von abnormalen Ablagerungen betroffen ist, ist die perforierende Keratoplastik (PKP) indiziert. Die Entscheidung für das Operationsverfahren basiert auf der präoperativen Endothelbeurteilung.
6. Pathophysiologie und detaillierter Pathogenesemechanismus
Das CHST6-Gen kodiert für die Kohlenhydrat-Sulfotransferase 6 3). Dieses Enzym ist für die Übertragung einer Sulfatgruppe auf N-Acetylglucosamin am Keratanmolekül verantwortlich und essentiell für die normale Synthese von Keratansulfat (KS) in den Hornhaut-Proteoglykanen.
Durch Genmutationen verursachter Verlust der Enzymaktivität führt zur Synthese von hyposulfatiertem Keratansulfat mit unzureichender Sulfatierung. Dieses abnormale Keratansulfat ist weniger löslich und lagert sich abnormal innerhalb und außerhalb der Keratozyten des Hornhautstromas ab 2,3).
Quantitative und qualitative Anomalien des Keratansulfats führen zu folgender Kaskade pathologischer Veränderungen.
Abnorme Produktion kleiner Proteoglykane (SLRP): Die für die Hornhaut spezifischen SLRP wie Lumican, Keratocan und Mimecan werden nicht normal synthetisiert.
Abnorme Anordnung der Kollagenfasern: Diese SLRP kontrollieren streng den Durchmesser und den Abstand der Kollagenfasern der Hornhaut und gewährleisten so die Transparenz. Eine verminderte Funktion der SLRP führt zu einem ungleichmäßigen Durchmesser der Kollagenfasern und einer Veränderung des Faserabstands2).
Abnorme Akkumulation in der extrazellulären Matrix: Nicht sulfatiertes Keratan selbst lagert sich in der extrazellulären Matrix ab.
Zunahme der Lichtstreuung und Verlust der Transparenz: Die oben genannten kombinierten Veränderungen erhöhen die Streuung des sichtbaren Lichts, und die gesamte Hornhaut wird diffus getrübt.
Die Akkumulation von Glykosaminoglykanen wird innerhalb und außerhalb der Stroma-Keratinozyten beobachtet, und mit fortschreitender Läsion breitet sie sich auf die Bowman-Membran, die Descemet-Membran und die Endothelzellen aus. Bei Typ I wurde auch im Ohrknorpel eine verminderte Enzymaktivität bestätigt, was darauf hindeutet, dass es sich um eine Teilmanifestation einer systemischen Störung des Keratansulfatstoffwechsels handeln könnte2). Klinisch treten systemische Symptome jedoch selten auf, und die Erkrankung wird als lokalisierte Erkrankung mit dem Hauptsymptom Hornhaut behandelt.
Unter den Hornhaut-Proteoglykanen kontrolliert Lumican den Durchmesser der Kollagenfasern auf etwa 25 nm, während Keratocan und Mimecan den Faserabstand gleichmäßig halten. Wenn die sulfatierten Seitenketten dieser SLRP kurz und unvollständig sind, weisen die Kollagenfasern eine Variation im Durchmesser und einen ungleichmäßigen Abstand auf. Infolgedessen nimmt die Lichtstreuung im Hornhautstroma zu, was klinisch als mattglasartige Trübung beobachtet wird.
Zur Aufrechterhaltung der Hornhauttransparenz ist klassisch die Gittertheorie von Maurer bekannt, die auf der „Auslöschung der Lichtinterferenz durch eine geordnete gitterartige Anordnung der Kollagenfasern“ beruht. Bei der MCD wird diese Gitterstruktur durch die SLRP-Anomalie gestört, was zum Verlust der Transparenz führt2,4).
Aktuelle Grundlagenforschung hat berichtet, dass eine Funktionsstörung der Autophagie aufgrund der CHST6-Mutation eine Pyroptose (inflammatorischer Zelltod) der Keratinozyten induzieren und so zum Fortschreiten der Erkrankung beitragen könnte8). Ähnliche Autophagie-Anomalien wurden auch bei anderen Hornhautdystrophien (wie dem granulären Typ II) berichtet und erregen als gemeinsame Pathologie aller Hornhautdystrophien Aufmerksamkeit.
Die gezielte Gentherapie wird als dauerhafte Behandlungsstrategie vorgeschlagen 8). Bei der Meesmann-Hornhautepitheldystrophie schreitet die Grundlagenforschung zur Genbearbeitung mit CRISPR/Cas9 voran, und könnte auch für die MCD eine zukünftige Behandlungsoption darstellen. Allerdings bleiben viele Herausforderungen für die klinische Anwendung bestehen, wie unbeabsichtigte Bearbeitung normaler Allele (Off-Target-Effekte), die Etablierung effizienter Gentransfermethoden in Hornhautstromazellen und die Überprüfung der Langzeitsicherheit.
Ein Ansatz zur enzymatischen Entfernung von akkumuliertem hyposulfatiertem Keratansulfat in der Hornhaut wird grundlegend untersucht. Es handelt sich um die lokale Anwendung des Konzepts der Enzymersatztherapie, das bereits bei systemischen Mukopolysaccharidosen führend ist, aber derzeit gibt es keine Berichte über eine klinische Anwendung.
Die Descemet-Membran-Stripping-Endothel-Keratoplastik (DSAEK) und die Descemet-Membran-Endothel-Keratoplastik (DMEK) sind Verfahren, die hauptsächlich für die Fuchs-Endotheldystrophie und die bullöse Keratopathie entwickelt wurden, aber ihre Anwendung bei MCD-Fällen mit vorherrschender Endothelbeteiligung ist ein zukünftiges Forschungsthema. Derzeit ist die perforierende Keratoplastik (PKP) die realistische Option für diese Erkrankung, die sowohl das Stroma als auch das Endothel betrifft. Zukünftig wurde das Konzept der „sequenziellen Keratoplastik“ vorgeschlagen, bei der die DALK (tiefe anteriore lamelläre Keratoplastik) zum alleinigen Ersatz des Stromas und die DMEK zum alleinigen Ersatz des Endothels schrittweise kombiniert werden, aber diese befinden sich noch im Forschungsstadium.
Der Serum-Keratansulfat-Spiegel soll bei MCD-Patienten mit Immunphänotyp II niedriger als normal sein, und sein Nutzen als systemischer metabolischer Marker wird diskutiert 2). Zukünftig wird eine Anwendung für das Screening mittels Blutuntersuchung und die frühe Trägerdiagnose in Familien mit CHST6-Mutationen erwartet. Darüber hinaus ist die automatisierte Analyse von Spaltlampenbildern durch künstliche Intelligenz ein Forschungsgebiet, das zur Früherkennung seltener Krankheiten wie dieser beitragen könnte.
Da es sich um eine autosomal-rezessive Erkrankung handelt, können etwa 25 % der Geschwister eines Patienten betroffen sein. Die CHST6-Gentestung ermöglicht ein Familien-Screening und eine genetische Beratung bezüglich Heirat und Schwangerschaft. Bei Familien mit Konsanguinität in der Vorgeschichte oder wenn Familienmitglieder ähnliche Hornhauttrübungen aufweisen, wird eine frühzeitige Überweisung an einen Spezialisten empfohlen.
Akama TO, Nishida K, Nakayama J, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nature Genetics. 2000;26(2):237-241. doi:10.1038/79987.
Aggarwal S, Peck T, Golen J, Karcioglu ZA. Macular corneal dystrophy: A review. Survey of Ophthalmology. 2018;63(5):609-617. doi:10.1016/j.survophthal.2018.03.004.
Jonasson F, Oshima E, Thonar EJ, et al. Macular corneal dystrophy in Iceland. A clinical, genealogic, and immunohistochemical study of 28 patients. Ophthalmology. 1996;103(7):1111-1117. doi:10.1016/s0161-6420(96)30559-9.
Jonasson F, Johannsson JH, Garner A, Rice NS. Macular corneal dystrophy in Iceland. Eye (London, England). 1989;3 ( Pt 4):446-54. doi:10.1038/eye.1989.66. PMID:2606219.
Sultana A, Sridhar MS, Jagannathan A, et al. Novel mutations of the carbohydrate sulfotransferase-6 (CHST6) gene causing macular corneal dystrophy in India. Mol Vis. 2003;9:730-734. PMID:14735064.
Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating keratoplasty for macular corneal dystrophy. Ophthalmology. 2005;112(2):220-224. doi:10.1016/j.ophtha.2004.08.017.
Reinhart WJ, Musch DC, Jacobs DS, Lee WB, Kaufman SC, Shtein RM. Deep anterior lamellar keratoplasty as an alternative to penetrating keratoplasty a report by the american academy of ophthalmology. Ophthalmology. 2011;118(1):209-218. doi:10.1016/j.ophtha.2010.11.002. PMID:21199711.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.