患病率的地区差异
美国:罕见,约每25万人0.3例 2,3)
冰岛:约每25万人19例,是全球最高频率的地区之一 5,6)
高患病率地区:南印度、沙特阿拉伯、冰岛和北欧频率较高 5,7)
其他地区:相对罕见。近亲结婚或复合杂合子发病

斑状角膜营养不良(macular corneal dystrophy:MCD)是一种遗传性角膜营养不良,其特征是糖胺聚糖(主要是硫酸角质素)在角膜基质中积聚。它呈常染色体隐性遗传,由位于第16号染色体长臂(16q22)的CHST6基因突变引起1,3)。曾被称为Groenouw角膜营养不良II型或Fehr角膜营养不良。
与许多其他角膜基质营养不良(颗粒状、格子状)为常染色体显性遗传不同,本病的特征是常染色体隐性遗传。在日本,它与颗粒状角膜营养不良(I型、II型)、格子状角膜营养不良(I型、IIIA型)和胶滴状角膜营养不良并称为四大角膜营养不良,约占所有角膜营养不良的96%。其中前两者为常染色体显性遗传,后两者(胶滴状和斑状)为常染色体隐性遗传。
在IC3D(国际角膜营养不良分类委员会)分类中,MCD被定位为一种基质营养不良 1)。Aggarwal等人在2018年《Survey of Ophthalmology》的综述中,将本病详述为“罕见但严重影响视功能的基质营养不良”,并总结了诊断和治疗体系 4)。在IC3D分类第二版中,角膜营养不良分为1至4类,根据致病基因、病理学发现和临床证据强度进行区分 1)。MCD因CHST6基因突变的鉴定被归类为第1类(基因水平已确立的营养不良)。
本病的歷史背景方面,1890年由Groenouw首次描述,后来出现了将颗粒状营养不良称为“I型”、斑状营养不良称为“II型”的惯例。1938年Jones和Zimmerman将其确立为独立疾病,2000年Akama等人鉴定出CHST6基因,阐明了其分子基础 3)。
全球范围内地区差异很大,患病率高的地区有家族聚集性。这是一种相对罕见的疾病。
患病率的地区差异
美国:罕见,约每25万人0.3例 2,3)
冰岛:约每25万人19例,是全球最高频率的地区之一 5,6)
高患病率地区:南印度、沙特阿拉伯、冰岛和北欧频率较高 5,7)
其他地区:相对罕见。近亲结婚或复合杂合子发病
免疫表型
MCD的免疫表型通过使用抗硫酸角质素单克隆抗体检测角膜和血清中的硫酸角质素量进行分类 2,3)。
| 表型 | 角膜硫酸角质素 | 血清硫酸角质素 |
|---|---|---|
| I型 | 阴性 | 阴性 |
| IA型 | 阳性(细胞内) | 阴性 |
| II型 | 阳性 | 阳性 |
大多数患者属于I型或IA型。然而,临床上区分这些亚型并不重要,且无法通过检查结果鉴别2,8)。
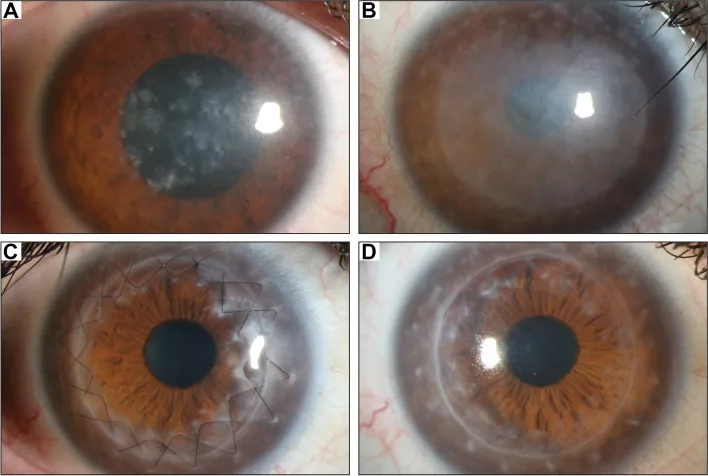
临床所见的典型进展模式如下。
临床上,角膜实质可见弥漫性细小沉积,呈磨玻璃样混浊。随着进展,混浊累及实质全层,并从中央向周边扩散。之后,除了淡薄混浊外,实质浅层至深层可见大量灰白色、形状不规则的小混浊。9)
初期所见
斑点状混浊:角膜中央部实质浅层出现灰白色小斑点状混浊
磨玻璃样混浊:角膜实质可见弥漫性淡薄混浊
边界不清:混浊边缘不清晰,与正常实质的边界模糊
进展期表现
全层受累:混浊累及整个基质层
向周边扩展:混浊从中央向周边扩散
角膜变薄:中央角膜厚度减少
内皮及Descemet膜沉积:深层结构也有异常物质积聚
裂隙灯显微镜下,整个角膜呈弥漫性混浊,其中可见灰白色不规则斑片状沉积物。用裂隙光做光学切面时,可见中央部沉积物位于浅层,周边部位于深层的特征性分布。斑片状病变多呈同心圆状出现8)。
混浊可累及角膜缘,这是与其他角膜营养不良的重要鉴别点。颗粒状角膜营养不良和格子状角膜营养不良的角膜缘常保持透明,而MCD的整个角膜包括角膜缘常发生混浊2,8)。不规则散光与前基质沉积有关,也可出现角膜知觉减退。由于内皮也有异常物质沉积,进展期病例可因内皮功能下降而并发基质水肿8)。
自然病程因人而异,但常经历以下阶段:
MCD是一种进行性疾病,一生中视功能持续下降,这与视力损害较轻的颗粒状角膜营养不良I型和部分格子状角膜营养不良亚型的预后显著不同4,8)。
致病基因是 CHST6(碳水化合物磺基转移酶6)3)。该基因位于16q22,编码一种将硫酸基团转移到角膜蛋白聚糖上N-乙酰葡糖胺的酶。该基因的突变类型非常多,包括错义突变、无义突变、移码突变和5’上游区域缺失等,已在不同民族中报道3,7)。
由于是常染色体隐性遗传,通常先证者的父母双方都是携带者。在近亲结婚较多的地区或群体中,发病率往往更高。不同家族间通婚产生的复合杂合子也可能发病7)。
Akama等人于2000年将CHST6鉴定为本病的致病基因,并表明免疫表型I型和II型均由同一基因座的突变引起3)。这一发现表明免疫表型的差异是由单个基因的不同突变模式决定的,是一项重要发现,为后续的基因诊断体系奠定了基础。
已报道的突变类型超过200种,其中错义突变最多。Sultana等人在南印度患者群中鉴定出大量新突变,表明该地区的高发病率是由于区域特异性突变的积累所致7)。沙特阿拉伯和冰岛也报道了类似的区域性聚集,认为人群的历史背景(奠基者效应和近亲结婚习俗)对发病率有贡献5,6,7)。
发病率因地区而异,是一种相对罕见的疾病9)。与颗粒状角膜营养不良等其他角膜营养不良相比,病例较少,且往往在具有复合杂合子或近亲结婚背景的家庭中报道。
怀疑MCD的线索是“双眼性、进行性、整个角膜弥漫性混浊的青春期至年轻成人”。首先详细询问自觉症状(视力下降、畏光、刺激感)的经过、家族史、近亲结婚史。然后进行裂隙灯显微镜下的角膜所见评估、内皮功能评估,必要时进行基因检测,这是标准流程。
这是诊断的基础检查。如果看到无充血或角膜水肿的双眼角膜混浊,应怀疑角膜营养不良。MCD具有以下特征性所见:
许多角膜营养不良在裂隙灯水平表现为不连续的病变(沉积物之间有透明部分),而MCD则例外地呈现弥漫性混浊模式。它与格子状角膜营养不良I型和胶样滴状角膜营养不良一起,被特征化为“弥漫性观察到的角膜沉积”的代表性例子。
CHST6基因分析有助于确诊。可在经认证的医疗机构进行。
组织学上,阿尔新蓝和胶体铁染色呈阳性,角膜基质内角膜细胞内外可见弥漫性低硫酸化糖胺聚糖积聚2,8)。可见Bowman层断裂,进展期病例内皮细胞内也可见异常物质。Descemet膜上也可出现滴状角膜(guttae)样表现。
| 疾病 | 遗传方式 | 特征 |
|---|---|---|
| 颗粒状角膜营养不良 | 常染色体显性 | 边界清晰的颗粒状沉积物(之间有透明区域) |
| 格子状角膜营养不良I型 | 常染色体显性 | 线状和网状格子线(淀粉样蛋白) |
| 胶样滴状角膜营养不良 | 常染色体隐性遗传 | 桑葚样或带状隆起性病变 |
| Schnyder结晶状角膜营养不良 | 常染色体显性遗传 | 针状结晶,合并高脂血症 |
| 先天性角膜基质营养不良 | 常染色体显性遗传 | 出生时即存在,基质增厚 |
| 全身性黏多糖贮积症(角膜型) | 隐性/X连锁遗传 | 伴有全身症状 |
此外,后部不定形角膜营养不良(PACD)和Descemet膜前角膜营养不良(PDCD)也需鉴别。全身性黏多糖贮积症(如Hurler、Scheie、Morquio综合征等)也可引起角膜混浊,因此需要结合全身表现进行评估8)。
MCD的治疗目标可归纳为三点:(1) 维持和恢复视功能,(2) 通过稳定眼表缓解疼痛和刺激症状,(3) 预防并发症(上皮糜烂、感染)。由于没有根本性的病因治疗,根据病程和症状进行阶段性干预。
目前尚无抑制本病进展的药物疗法8)。以下措施用于缓解症状。
对于视力进行性下降的病例,角膜移植术是唯一的根治手段。根据有无内皮病变选择术式。
在MCD中,内皮细胞也可能有异常沉积,因此当病变累及深层时,倾向于选择PKP而非DALK7)。伴有内皮异常时,全层角膜移植是适应症。术前详细的内皮评估(镜面显微镜和共聚焦显微镜)是决定术式的关键。
DALK后的复发率较低,据报道约为5%。PKP后的植片存活率良好,许多报告显示可获得长期存活,但也有报告称术后数年至十多年后异常物质会重新沉积在植片上8)。Al-Swailem等人在沙特阿拉伯的系列研究中,MCD患者PKP后的植片存活率良好,但部分病例在长期随访中观察到复发8)。此外,Reinhart等人的AAO综述显示,对于实质层营养不良,DALK可实现与PKP相当或更好的视力恢复,并同时降低内皮细胞丢失率9)。Unal等人的多中心研究也报告了DALK对包括MCD在内的实质层营养不良的有效性。
一般来说,角膜植片术后复发是由于原发病的分子病理状态残留在宿主侧所致。DALK保留了宿主的内皮和Descemet膜前层,因此在内皮受累的病例中,病变可能进展,需注意。术后需要长期定期观察(视力、裂隙灯检查、内皮细胞密度测量)。
CHST6基因编码碳水化合物磺基转移酶6 3)。该酶负责将硫酸基团转移到角质素分子上的N-乙酰葡糖胺,是角膜蛋白聚糖中硫酸角质素(KS)正常合成所必需的。
基因突变导致酶活性丧失时,会合成硫酸化不足的低硫酸化角质素硫酸盐。这种异常的角质素硫酸盐溶解度低,在角膜基质中的角膜细胞内外异常沉积2,3)。
硫酸角质素的量和质异常会导致以下连锁病理变化。
糖胺聚糖的积聚在基质角膜细胞的细胞内和细胞外均可观察到,随着病变进展,可扩散至Bowman层、Descemet膜和内皮细胞。在I型中,耳廓软骨中也观察到酶活性降低,提示可能是全身性硫酸角质素代谢异常的部分表现2)。但临床上全身症状少见,通常作为以角膜为主要症状的局限性疾病处理。
在角膜蛋白聚糖中,lumican负责将胶原纤维直径控制在约25 nm,而keratocan和mimecan维持纤维间距均匀。当这些SLRP的硫酸化侧链短而不完整时,胶原纤维粗细不一,间距也不均匀。结果导致角膜基质内光散射增加,临床表现为磨玻璃样混浊。
角膜透明性的维持经典地由Maurer格子理论解释,即胶原纤维整齐的格子状排列通过光干涉相消实现。在MCD中,这种格子结构因SLRP异常而破坏,导致透明性丧失2,4)。
近年来的基础研究报告称,CHST6突变导致的自噬功能障碍可能诱导角膜细胞焦亡(炎症性细胞死亡),促进疾病进展8)。其他角膜营养不良(如颗粒状II型)中也报道了类似的自噬异常,作为角膜营养不良的共同病理机制受到关注。
基因靶向治疗已被提出作为一种永久性治疗策略8)。在Meesmann角膜上皮营养不良中,使用CRISPR/Cas9进行基因编辑的基础研究正在推进,未来也可能成为MCD的治疗选择。然而,临床应用中仍存在许多挑战,包括对正常等位基因的意外编辑(脱靶效应)、建立向角膜基质细胞高效递送基因的方法以及长期安全性的验证。
一种通过酶学方法去除角膜内积聚的低硫酸化硫酸角质素的方法正在基础研究中。这借鉴了全身性黏多糖贮积症中已应用的酶替代疗法概念,并将其局部应用于角膜,但目前尚无临床应用的报道。
Descemet膜剥离角膜内皮移植术(DSAEK)和Descemet膜角膜内皮移植术(DMEK)主要是针对Fuchs角膜内皮营养不良和大疱性角膜病变发展起来的手术方式,但将其应用于MCD内皮病变先行病例也是未来的研究课题。目前,对于这种同时累及基质和内皮的疾病,穿透性角膜移植术(PKP)是现实的选择。未来,有学者提出了“序贯角膜移植术”的概念,即分阶段联合使用仅置换基质的深板层角膜移植术(DALK)和仅置换内皮的DMEK,但这些仍处于研究阶段。
据报道,免疫表型II型MCD患者的血清硫酸角质素水平低于正常值,其作为全身代谢标志物的有用性正在讨论中2)。未来有望应用于血液筛查和CHST6突变家系的早期携带者诊断。此外,利用人工智能自动分析裂隙灯图像也是可能有助于此类罕见病早期发现的研究领域。
由于该病为常染色体隐性遗传,患者同胞中约有25%可能发病。利用CHST6基因检测,可以进行家族内筛查以及关于婚姻和妊娠的遗传咨询。对于有近亲结婚史或家族内有类似角膜混浊病例的家系,建议早期转诊至专科医生。