La dystrophie maculaire de la cornée (macular corneal dystrophy : MCD) est une dystrophie cornéenne héréditaire caractérisée par l’accumulation de glycosaminoglycanes (principalement du sulfate de kératane) dans le stroma cornéen. Elle est transmise selon un mode autosomique récessif et est causée par une mutation du gène CHST6 situé sur le bras long du chromosome 16 (16q22)1,3). Autrefois, elle était également appelée dystrophie cornéenne de Groenouw de type II ou dystrophie cornéenne de Fehr.
Contrairement à de nombreuses autres dystrophies du stroma cornéen (granulaire, grillagée) qui sont autosomiques dominantes, cette maladie se caractérise par une transmission autosomique récessive. Au Japon, elle est considérée comme l’une des quatre grandes dystrophies cornéennes avec la dystrophie granulaire (types I et II), la dystrophie grillagée (types I et IIIA) et la dystrophie gélatineuse en gouttes, représentant environ 96 % de toutes les dystrophies cornéennes. Parmi celles-ci, les deux premières sont autosomiques dominantes, tandis que les deux dernières (gélatineuse en gouttes et maculaire) sont autosomiques récessives.
Selon la classification IC3D (International Committee for Classification of Corneal Dystrophies), la MCD est classée comme un type de dystrophie stromale 1). Dans une revue de 2018 dans Survey of Ophthalmology, Aggarwal et al. décrivent cette maladie comme « une dystrophie stromale rare mais ayant un impact significatif sur la fonction visuelle » et résument le système de diagnostic et de traitement 4). Dans la deuxième édition de la classification IC3D, les dystrophies cornéennes sont classées en catégories 1 à 4, en fonction de la force des preuves concernant le gène causal, les résultats pathologiques et le tableau clinique 1). La MCD est classée dans la catégorie 1 (dystrophie établie au niveau génétique) grâce à l’identification de mutations du gène CHST6.
Historiquement, cette maladie a été décrite pour la première fois par Groenouw en 1890, et plus tard, la dystrophie granuleuse a été appelée « type I » et la dystrophie maculaire « type II ». En 1938, Jones et Zimmerman l’ont établie comme une maladie indépendante, et en 2000, Akama et al. ont identifié le gène CHST6, clarifiant ainsi la base moléculaire 3).
Il existe des variations régionales importantes dans le monde, et dans les zones à forte prévalence, on observe une accumulation familiale. C’est une maladie relativement rare.
Variations régionales de la prévalence
États-Unis : environ 0,3 personne pour 250 000 habitants, rare 2,3)
Islande : environ 19 personnes pour 250 000 habitants, l’une des régions les plus fréquentes au monde 5,6)
Régions à forte prévalence : fréquence élevée dans le sud de l’Inde, l’Arabie saoudite, l’Islande et les pays nordiques 5,7)
Autres régions : relativement rare. Survient en cas de mariage consanguin ou d’hétérozygotie composite
Immunophénotype
Type I : kératane sulfate négatif dans la cornée et le sérum 2)
Type IA : positif dans les kératocytes cornéens, négatif dans le sérum 2)
Type II : kératane sulfate positif dans la cornée et le sérum 2)
Tableau clinique : les trois types ont le même phénotype et ne peuvent pas être distingués à la lampe à fente2,8)
L’immunophénotype de la MCD est classé en fonction de la quantité de kératane sulfate dans la cornée et le sérum à l’aide d’anticorps monoclonaux anti-kératane sulfate 2,3).
Phénotype
Kératane sulfate cornéen
Kératane sulfate sérique
Type I
Négatif
Négatif
Type IA
Positif (intracellulaire)
Négatif
Type II
Positif
Positif
La plupart des patients sont classés comme type I ou IA. Cependant, cliniquement, la distinction entre ces sous-types n’est pas importante et ils ne peuvent pas être différenciés par l’examen clinique2,8).
QEn quoi la dystrophie cornéenne maculaire est-elle différente des autres dystrophies cornéennes ?
A
La principale différence est qu’elle suit un mode de transmission autosomique récessif. Les dystrophies granulaires et grillagées sont autosomiques dominantes, mais cette maladie nécessite des mutations sur les deux allèles du gène CHST6. De plus, elle se présente avec une opacité diffuse en verre dépoli, un blanchiment de toute la cornée, et à la lampe à fente, les dépôts sont superficiels au centre et profonds en périphérie.
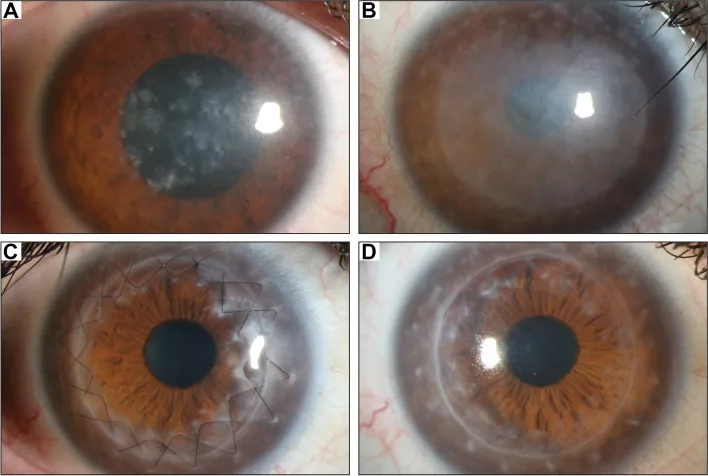
Gassel CJ, et al. Histological findings of corneal tissue after failed phototherapeutic keratectomy in macular corneal dystrophy - a case report. BMC Ophthalmol. 2022. Figure 1. PMCID: PMC9080147. License: CC BY.
Photographie à la lampe à fente montrant une opacité diffuse grisâtre et des dépôts maculaires du centre à l’ensemble de la cornée. Cela illustre les signes cliniques typiques de la dystrophie cornéenne maculaire, facilitant la compréhension de l’opacité cornéenne responsable de la baisse de vision.
Baisse de vision : plainte la plus fréquente. Souvent remarquée entre 10 et 30 ans8)
Douleur oculaire et sensation d’irritation : peut survenir avec des érosions épithéliales récurrentes
Photophobie (sensibilité à la lumière) : la fonction visuelle diminue considérablement dans les environnements lumineux à mesure que l’opacité cornéenne progresse
Érosions épithéliales récurrentes : peuvent survenir de manière répétée en raison d’une adhésion anormale de l’épithélium cornéen
Le schéma de progression typique des signes cliniques est le suivant.
Cliniquement, de fins dépôts diffus sont observés dans le stroma cornéen, donnant un aspect de verre dépoli. L’opacité progresse pour impliquer toute l’épaisseur du stroma et s’étend du centre vers la périphérie. Par la suite, en plus de l’opacité légère, de nombreuses petites opacités grisâtres de forme irrégulière apparaissent dans les couches superficielles à profondes du stroma.9)
Signes précoces
Opacités ponctuées : de petites opacités ponctuées grisâtres apparaissent dans les couches superficielles du stroma au centre de la cornée
Opacité en verre dépoli : opacité légère diffuse du stroma cornéen
Limites floues : les bords de l’opacité sont indistincts, avec une limite peu claire entre le stroma normal et anormal
Signes de stade avancé
Extension à toute l’épaisseur : l’opacité atteint toute l’épaisseur du stroma
Extension vers la périphérie : l’opacité s’étend du centre vers la périphérie
Amincissement cornéen : l’épaisseur de la cornée centrale diminue
Dépôts sur l’endothélium et la membrane de Descemet : des substances anormales s’accumulent également dans les structures profondes
À la lampe à fente, la cornée entière est diffusément opaque, avec des dépôts irréguliers gris-blanc. En coupe optique, les dépôts sont superficiels au centre et profonds en périphérie, une distribution caractéristique. Les lésions apparaissent souvent en motifs concentriques8).
L’opacité peut s’étendre au limbe, un point clé pour distinguer la MCD des autres dystrophies cornéennes. Dans la dystrophie granuleuse et la dystrophie réticulée, le limbe reste souvent clair, alors que dans la MCD, la cornée entière jusqu’au limbe est souvent opaque2,8). L’astigmatisme irrégulier est lié aux dépôts stromaux antérieurs, et une hypoesthésie cornéenne peut survenir. Les dépôts endothéliaux peuvent entraîner un œdème stromal par dysfonction endothéliale dans les cas avancés8).
L’évolution naturelle varie selon les individus, mais suit souvent les étapes suivantes.
Petite enfance (stade asymptomatique) : la mutation génétique est présente dès la naissance, mais les signes à la lampe à fente sont rares et le patient est asymptomatique
Âge scolaire à adolescence (stade d’opacité précoce) : une opacité diffuse légère apparaît dans le stroma antérieur, puis des dépôts maculaires sont observés
10 à 30 ans (stade de baisse visuelle) : l’opacité progresse et le patient prend conscience de la baisse de vision
30 à 40 ans (stade avancé) : l’opacité s’étend à toute l’épaisseur du stroma et au limbe, avec hypoesthésie cornéenne, amincissement et astigmatisme irrégulier
Âge moyen à avancé (stade d’indication de greffe de cornée) : la fonction visuelle est suffisamment réduite pour interférer avec la vie quotidienne, et une greffe est envisagée
La MCD est une maladie progressive avec un déclin continu de la fonction visuelle tout au long de la vie, ce qui la distingue des dystrophies granuleuse de type I et réticulée (certains sous-types) où la déficience visuelle reste légère4,8).
Le gène responsable est CHST6 (carbohydrate sulfotransferase 6)3). Situé en 16q22, il code une enzyme qui transfère un groupe sulfate à la N-acétylglucosamine sur les protéoglycanes cornéens. Les mutations de ce gène sont très nombreuses : mutations faux-sens, non-sens, décalages du cadre de lecture, délétions dans la région 5’ non traduite, rapportées dans diverses populations3,7).
La transmission est autosomique récessive, donc les deux parents du proposant sont généralement porteurs. La fréquence est plus élevée dans les régions ou groupes où les mariages consanguins sont fréquents. Les hétérozygotes composites issus de mariages entre familles différentes peuvent également développer la maladie7).
Akama et al. ont identifié CHST6 comme le gène responsable en 2000, montrant que les immunophénotypes I et II sont tous deux causés par des mutations du même locus3). Cette découverte a suggéré que les différences d’immunophénotype sont déterminées par différents types de mutations d’un seul gène, jetant les bases du diagnostic génétique ultérieur.
Plus de 200 mutations ont été rapportées, les mutations faux-sens étant les plus fréquentes. Sultana et al. ont identifié de nombreuses nouvelles mutations chez des patients du sud de l’Inde, montrant que la fréquence élevée dans cette région résulte d’une accumulation de mutations spécifiques à la population7). Des accumulations régionales similaires ont été rapportées en Arabie saoudite et en Islande, suggérant que l’histoire de la population (effet fondateur, coutumes de mariages consanguins) contribue à la fréquence de la maladie5,6,7).
La fréquence varie selon les régions ; c’est une maladie relativement rare9). Comparée à d’autres dystrophies cornéennes comme la dystrophie granuleuse, elle est moins fréquente et rapportée surtout dans les familles avec hétérozygotie composite ou antécédents de consanguinité.
Antécédents familiaux : transmission autosomique récessive, les deux parents doivent être porteurs
Consanguinité : augmente l’incidence
Région géographique : prévalence élevée en Inde du Sud, Arabie saoudite, Islande, Scandinavie5,7)
QLe test génétique est-il nécessaire ?
A
Le test du gène CHST6 est utile pour confirmer le diagnostic. Il peut être réalisé dans des établissements agréés. Cependant, le diagnostic clinique à la lampe à fente reste prédominant dans de nombreux centres. Le test est utile pour évaluer le risque chez les futurs enfants ou pour confirmer le diagnostic dans les cas atypiques.
La suspicion de MCD repose sur la triade : bilatéralité, progression, opacité cornéenne diffuse chez l’adolescent ou le jeune adulte. On commence par recueillir l’évolution des symptômes subjectifs (baisse de vision, photophobie, irritation), les antécédents familiaux et la consanguinité. Ensuite, l’examen standard comprend l’évaluation de la cornée à la lampe à fente, l’évaluation de la fonction endothéliale et, si nécessaire, un test génétique.
C’est l’examen de base du diagnostic. En présence d’une opacité cornéenne bilatérale sans hyperhémie ni œdème cornéen, il faut suspecter une dystrophie cornéenne. La MCD présente les caractéristiques suivantes :
Opacité diffuse en verre dépoli : la cornée entière est trouble de manière diffuse
Dépôts maculaires : nombreuses opacités irrégulières blanc-gris
Distribution concentrique : à la coupe optique, les dépôts sont superficiels au centre et profonds en périphérie
Infiltration limbique : l’opacité peut s’étendre jusqu’au limbe
Anomalies endothéliales : dans les cas avancés, on peut observer des dépôts en gouttes
Alors que la plupart des dystrophies cornéennes apparaissent comme des lésions discontinues (avec des zones transparentes entre les dépôts) au niveau de la lampe à fente, la MCD présente exceptionnellement un motif d’opacité diffuse. Elle est caractérisée comme l’un des exemples typiques de « dépôts cornéens observés de manière diffuse » avec la dystrophie cornéenne grillagée de type I et la dystrophie cornéenne gélatineuse en gouttes.
Microscopie confocale in vivo : montre un matériel hyperréflectif aux limites floues et une disparition des kératocytes normaux8)
Topographie cornéenne : montre une densité accrue au sommet de la cornée et un amincissement cornéen central
Microscope ultrasonique à balayage (UBM) : utile pour évaluer les opacités profondes et la structure postérieure de la cornée
Microscope spéculaire : évaluation de la densité et de la morphologie des cellules endothéliales. Le degré de pathologie endothéliale influence directement le choix de la technique chirurgicale
Pentacam cornéen (appareil de photographie Scheimpflug) : permet d’obtenir une carte de densité de toute l’épaisseur cornéenne, utile pour l’évaluation tridimensionnelle des zones d’opacité
Histologiquement, la coloration au bleu Alcian et la coloration au fer colloïdal sont positives, et on observe une accumulation diffuse de glycosaminoglycanes hyposulfatés à l’intérieur et à l’extérieur des kératocytes du stroma cornéen2,8). Des ruptures de la membrane de Bowman peuvent être observées, et dans les cas avancés, des substances anormales sont également présentes dans les cellules endothéliales. Des aspects de gouttelettes (guttae) peuvent également être observés sur la membrane de Descemet.
Dépôts granulaires bien délimités (avec zones transparentes entre eux)
Dystrophie cornéenne grillagée de type I
Autosomique dominant
Lignes grillagées linéaires et réticulaires (amyloïde)
Dystrophie cornéenne en gouttes gélatineuses
Autosomique récessive
Lésions surélevées en mûrier ou en bande
Dystrophie cristalline de Schnyder
Autosomique dominante
Cristaux en aiguille, associée à une hyperlipidémie
Dystrophie cornéenne stromale congénitale
Autosomique dominante
Présente dès la naissance, épaississement stromal
Mucopolysaccharidose systémique (type cornéen)
Récessive / liée à l’X
Avec symptômes systémiques
De plus, la dystrophie cornéenne postérieure polymorphe (PPMD) et la dystrophie cornéenne pré-Descemet (PDCD) sont également à considérer dans le diagnostic différentiel. Les mucopolysaccharidoses systémiques (syndromes de Hurler, Scheie, Morquio, etc.) peuvent également provoquer une opacité cornéenne, il est donc nécessaire d’évaluer les signes systémiques8).
QComment diagnostique-t-on la dystrophie maculaire de la cornée ?
A
Le diagnostic clinique repose sur l’examen à la lampe à fente montrant une opacité cornéenne diffuse en verre dépoli et des dépôts maculaires, une atteinte bilatérale et progressive, des antécédents familiaux et l’âge de début (10-30 ans). Le test génétique du gène CHST6 est utile pour confirmer le diagnostic.
Les objectifs du traitement de la MCD sont : (1) maintenir/récupérer la fonction visuelle, (2) soulager la douleur et les symptômes d’irritation en stabilisant la surface oculaire, et (3) prévenir les complications (érosion épithéliale, infection). Comme il n’existe pas de traitement étiologique curatif, une intervention progressive est réalisée en fonction du stade et des symptômes.
Aucun traitement médicamenteux n’a été établi pour ralentir la progression de cette maladie8). Les mesures suivantes sont prises pour soulager les symptômes :
Larmes artificielles : protection de la surface oculaire et prévention de la sécheresse
Dans les cas de baisse visuelle progressive, la greffe de cornée est le seul traitement curatif. La technique chirurgicale est choisie en fonction de la présence ou non d’une atteinte endothéliale.
Kératoplastie lamellaire antérieure profonde (DALK) : traitement de première intention pour les cas sans atteinte endothéliale8,9). En préservant l’endothélium cornéen du patient, le risque de rejet du greffon est plus faible qu’avec une kératoplastie transfixiante. Selon un rapport d’évaluation de l’AAO (Académie américaine d’ophtalmologie), la DALK permet une récupération visuelle équivalente à la PKP pour les dystrophies stromales, avec une perte endothéliale moindre8). Le taux de récidive après DALK est faible, et le remplacement du stroma cornéen du receveur rend la récidive peu probable.
Kératoplastie transfixiante (PKP) : indiquée dans les cas avancés avec dépôts de substance anormale dans l’endothélium ou la membrane de Descemet, ainsi que dans les cas d’amincissement cornéen central sévère7). L’âge moyen de la première PKP pour MCD se situe entre 30 et 40 ans7), et le taux de survie du greffon est bon.
Kératectomie photothérapeutique (PTK) : traitement symptomatique des érosions épithéliales récurrentes et des opacités cicatricielles superficielles. Il faut toutefois être attentif à l’induction d’hypermétropie et d’opacité stromale.
Étant donné que des substances anormales peuvent également se déposer dans les cellules endothéliales dans la MCD, la PKP a tendance à être préférée à la DALK lorsque la maladie s’étend profondément7). En cas d’anomalie endothéliale, une kératoplastie transfixiante est indiquée. Une évaluation endothéliale préopératoire détaillée (microscopie spéculaire, confocale) est essentielle pour déterminer la technique chirurgicale.
Le taux de récidive après DALK est faible, estimé à environ 5 %. Le taux de survie du greffon après PKP est bon, avec une survie à long terme rapportée dans de nombreuses études, bien que des cas de redépôt de substance anormale sur le greffon plusieurs années à plusieurs décennies après la chirurgie aient été signalés 8). Dans la série saoudienne d’Al-Swailem et al., le taux de survie après PKP pour la MCD était bon, mais une récidive à long terme a été observée dans certains cas 8). De plus, dans la revue de l’AAO par Reinhart et al., il a été montré que la DALK pour les dystrophies stromales offre une récupération visuelle égale ou supérieure à la PKP avec un faible taux de perte endothéliale 9). Une étude multicentrique d’Unal et al. a également rapporté l’efficacité de la DALK pour les dystrophies stromales, y compris la MCD.
En général, la récidive postopératoire des greffons cornéens survient parce que la pathologie moléculaire de la maladie d’origine persiste chez l’hôte. Dans la DALK, l’endothélium et la couche pré-descemétique de l’hôte sont préservés, il faut donc noter que la maladie peut progresser dans les cas où la pathologie endothéliale est présente. Une surveillance régulière à long terme (acuité visuelle, lampe à fente, mesure de la densité des cellules endothéliales) est nécessaire après la chirurgie.
QFaut-il choisir la DALK ou la PKP ?
A
Si la pathologie n’affecte pas l’endothélium ou la membrane de Descemet, la kératoplastie lamellaire antérieure profonde (DALK) est le premier choix. La DALK préserve l’endothélium cornéen du patient, ce qui réduit le risque de rejet, et le taux de récidive postopératoire est d’environ 5 %. En revanche, dans les cas où des dépôts anormaux affectent également l’endothélium, la kératoplastie transfixiante (PKP) est indiquée. La décision de la technique chirurgicale est basée sur l’évaluation endothéliale préopératoire.
6. Physiopathologie et mécanisme détaillé de la pathogenèse
Le gène CHST6 code pour la carbohydrate sulfotransférase 6 3). Cette enzyme est responsable du transfert d’un groupe sulfate vers la N-acétylglucosamine sur la molécule de kératane, et est essentielle à la synthèse normale du kératane sulfate (KS) présent dans les protéoglycanes cornéens.
La perte d’activité enzymatique due à une mutation génétique conduit à la synthèse d’un kératane sulfate hyposulfaté, moins soluble. Ce kératane sulfate anormal se dépose à l’intérieur et à l’extérieur des kératocytes du stroma cornéen2,3).
Les anomalies quantitatives et qualitatives du kératane sulfate entraînent la cascade pathologique suivante.
Production anormale de petits protéoglycanes riches en leucine (SLRP) : Les SLRP spécifiques de la cornée tels que le lumican, le kératocane et le mimécane ne sont plus synthétisés normalement.
Disposition anormale des fibres de collagène : Ces SLRP contrôlent strictement le diamètre et l’espacement des fibres de collagène cornéennes, assurant la transparence. La diminution de la fonction des SLRP entraîne une hétérogénéité du diamètre des fibres de collagène et une modification de leur espacement2).
Accumulation anormale dans la matrice extracellulaire : Le kératane non sulfaté lui-même se dépose dans la matrice extracellulaire.
Augmentation de la diffusion de la lumière et perte de transparence : Les changements combinés ci-dessus augmentent la diffusion de la lumière visible, rendant la cornée globalement trouble de manière diffuse.
L’accumulation de glycosaminoglycanes est observée à l’intérieur et à l’extérieur des kératocytes du stroma, et à mesure que la lésion progresse, elle s’étend à la membrane de Bowman, à la membrane de Descemet et aux cellules endothéliales. Dans le type I, une diminution de l’activité enzymatique a également été confirmée dans le cartilage de l’oreille, ce qui suggère qu’il pourrait s’agir d’une manifestation partielle d’un trouble systémique du métabolisme du sulfate de kératane2). Cependant, les symptômes systémiques sont cliniquement rares, et la maladie est traitée comme une affection localisée dont le principal symptôme est cornéen.
Parmi les protéoglycanes cornéens, le lumican contrôle le diamètre des fibres de collagène à environ 25 nm, tandis que le kératocane et le mimécane maintiennent un espacement uniforme des fibres. Lorsque les chaînes latérales sulfatées de ces SLRP sont courtes et incomplètes, les fibres de collagène présentent une variation de diamètre et un espacement irrégulier. En conséquence, la diffusion de la lumière dans le stroma cornéen augmente, ce qui est cliniquement observé comme une opacité en verre dépoli.
La théorie classique de Maurer sur le maintien de la transparence cornéenne repose sur « l’annulation de l’interférence lumineuse par un arrangement ordonné en treillis des fibres de collagène ». Dans la MCD, cette structure en treillis est perturbée par l’anomalie des SLRP, entraînant une perte de transparence2,4).
Des recherches fondamentales récentes ont rapporté que le dysfonctionnement de l’autophagie dû à la mutation CHST6 pourrait induire une pyroptose (mort cellulaire inflammatoire) des kératocytes, contribuant à la progression de la maladie8). Des anomalies similaires de l’autophagie ont été rapportées dans d’autres dystrophies cornéennes (comme le type granulaire II), et elles attirent l’attention en tant que pathologie commune à l’ensemble des dystrophies cornéennes.
La thérapie génique ciblée est proposée comme stratégie de traitement permanent 8). Des recherches fondamentales sur l’édition génétique utilisant CRISPR/Cas9 sont en cours pour la dystrophie épithéliale cornéenne de Meesmann, et pourraient également devenir une option thérapeutique future pour la MCD. Cependant, de nombreux défis subsistent pour l’application clinique, notamment les modifications non intentionnelles des allèles normaux (effets hors cible), l’établissement de méthodes efficaces de transfert de gènes dans les kératocytes cornéens, et la validation de la sécurité à long terme.
Une approche visant à éliminer enzymatiquement le sulfate de kératane hyposulfaté accumulé dans la cornée est étudiée au niveau fondamental. Il s’agit d’appliquer localement à la cornée le concept de thérapie enzymatique substitutive déjà utilisé dans les mucopolysaccharidoses systémiques, mais aucun rapport d’application clinique n’existe à ce jour.
La kératoplastie endothéliale par stripping de la membrane de Descemet (DSAEK) et la kératoplastie endothéliale de la membrane de Descemet (DMEK) sont des techniques développées principalement pour la dystrophie endothéliale de Fuchs et la kératopathie bulleuse, mais leur application aux cas de MCD avec atteinte endothéliale prédominante est un sujet de recherche futur. Actuellement, la kératoplastie pénétrante (PKP) reste l’option réaliste pour cette maladie qui affecte à la fois le stroma et l’endothélium. À l’avenir, le concept de « kératoplastie séquentielle » combinant la DALK (kératoplastie lamellaire antérieure profonde) pour remplacer uniquement le stroma et la DMEK pour remplacer uniquement l’endothélium a été proposé, mais ces approches en sont encore au stade de la recherche.
Le taux sérique de sulfate de kératane serait inférieur à la normale chez les patients atteints de MCD de type immunophénotype II, et son utilité en tant que marqueur métabolique systémique est discutée 2). À l’avenir, on espère une application au dépistage par analyse sanguine et au diagnostic précoce des porteurs dans les familles porteuses de mutations CHST6. De plus, l’analyse automatisée des images de la lampe à fente par intelligence artificielle est un domaine de recherche qui pourrait contribuer à la détection précoce de maladies rares comme celle-ci.
Étant donné qu’il s’agit d’une maladie autosomique récessive, environ 25 % des frères et sœurs d’un patient peuvent être atteints. Le test génétique CHST6 permet le dépistage familial et le conseil génétique concernant le mariage et la grossesse. Une orientation précoce vers un spécialiste est recommandée dans les familles avec des antécédents de consanguinité ou lorsque des membres de la famille présentent des opacités cornéennes similaires.
Akama TO, Nishida K, Nakayama J, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nature Genetics. 2000;26(2):237-241. doi:10.1038/79987.
Aggarwal S, Peck T, Golen J, Karcioglu ZA. Macular corneal dystrophy: A review. Survey of Ophthalmology. 2018;63(5):609-617. doi:10.1016/j.survophthal.2018.03.004.
Jonasson F, Oshima E, Thonar EJ, et al. Macular corneal dystrophy in Iceland. A clinical, genealogic, and immunohistochemical study of 28 patients. Ophthalmology. 1996;103(7):1111-1117. doi:10.1016/s0161-6420(96)30559-9.
Jonasson F, Johannsson JH, Garner A, Rice NS. Macular corneal dystrophy in Iceland. Eye (London, England). 1989;3 ( Pt 4):446-54. doi:10.1038/eye.1989.66. PMID:2606219.
Sultana A, Sridhar MS, Jagannathan A, et al. Novel mutations of the carbohydrate sulfotransferase-6 (CHST6) gene causing macular corneal dystrophy in India. Mol Vis. 2003;9:730-734. PMID:14735064.
Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating keratoplasty for macular corneal dystrophy. Ophthalmology. 2005;112(2):220-224. doi:10.1016/j.ophtha.2004.08.017.
Reinhart WJ, Musch DC, Jacobs DS, Lee WB, Kaufman SC, Shtein RM. Deep anterior lamellar keratoplasty as an alternative to penetrating keratoplasty a report by the american academy of ophthalmology. Ophthalmology. 2011;118(1):209-218. doi:10.1016/j.ophtha.2010.11.002. PMID:21199711.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.