Макулярная дистрофия роговицы (macular corneal dystrophy: MCD) — это наследственная дистрофия роговицы, характеризующаяся накоплением гликозаминогликанов (в основном кератансульфата) в строме роговицы. Она наследуется по аутосомно-рецессивному типу и вызывается мутацией гена CHST6, расположенного на длинном плече хромосомы 16 (16q22)1,3). Ранее она также называлась дистрофией роговицы Гроэноу II типа или дистрофией роговицы Фера.
В отличие от многих других стромальных дистрофий роговицы (гранулярной, решетчатой), которые являются аутосомно-доминантными, данное заболевание характеризуется аутосомно-рецессивным типом наследования. В Японии она считается одной из четырех основных дистрофий роговицы наряду с гранулярной (типы I, II), решетчатой (типы I, IIIA) и желатинозной каплевидной дистрофией, составляя около 96% всех дистрофий роговицы. Из них первые две являются аутосомно-доминантными, а последние две (желатинозная каплевидная и макулярная) — аутосомно-рецессивными.
Согласно классификации IC3D (Международный комитет по классификации дистрофий роговицы), MCD относится к типу стромальной дистрофии 1). В обзоре Aggarwal и соавт. в Survey of Ophthalmology за 2018 год это заболевание описывается как «редкая, но значительно влияющая на зрительную функцию стромальная дистрофия», и обобщается система диагностики и лечения 4). Во втором издании классификации IC3D дистрофии роговицы подразделяются на категории 1–4 в зависимости от силы доказательств, касающихся причинного гена, патологических находок и клинической картины 1). MCD классифицируется как категория 1 (дистрофия, установленная на генетическом уровне) благодаря идентификации мутаций гена CHST6.
Исторически это заболевание было впервые описано Groenouw в 1890 году, и позже возникла практика называть гранулярную дистрофию «типом I», а макулярную дистрофию — «типом II». В 1938 году Jones и Zimmerman установили его как самостоятельное заболевание, а в 2000 году Akama и соавт. идентифицировали ген CHST6, прояснив молекулярную основу 3).
Во всем мире наблюдаются значительные региональные различия; в регионах с высокой распространенностью отмечается семейное накопление. Это относительно редкое заболевание.
Региональные различия в распространенности
США : около 0,3 человека на 250 000 населения, редко 2,3)
Исландия : около 19 человек на 250 000 населения, один из самых частых регионов в мире 5,6)
Регионы с высокой распространенностью : высокая частота в Южной Индии, Саудовской Аравии, Исландии и скандинавских странах 5,7)
Другие регионы : относительно редко. Возникает при кровном родстве или компаунд-гетерозиготности
Иммунофенотип
Тип I : кератансульфат отрицательный в роговице и сыворотке 2)
Тип IA : положительный в кератоцитах роговицы, отрицательный в сыворотке 2)
Тип II : кератансульфат положительный в роговице и сыворотке 2)
Клиническая картина : все три типа имеют одинаковый фенотип и неразличимы при щелевой лампе 2,8)
Иммунофенотип MCD классифицируется по количеству кератансульфата в роговице и сыворотке с использованием моноклональных антител против кератансульфата 2,3).
Фенотип
Роговичный кератансульфат
Сывороточный кератансульфат
I тип
Отрицательный
Отрицательный
IA тип
Положительный (внутриклеточный)
Отрицательный
II тип
Положительный
Положительный
Большинство пациентов относятся к I или IA типу. Однако клинически различие между этими подтипами не важно, и их нельзя дифференцировать по данным осмотра2,8).
QЧем макулярная дистрофия роговицы отличается от других дистрофий роговицы?
A
Самое большое отличие — аутосомно-рецессивный тип наследования. Гранулярная и решетчатая дистрофии роговицы являются аутосомно-доминантными, но для этого заболевания необходимы мутации в обоих аллелях гена CHST6. Также она проявляется диффузным помутнением по типу матового стекла, побелением всей роговицы, а при осмотре в щелевой лампе отложения в центре поверхностные, а на периферии глубокие.
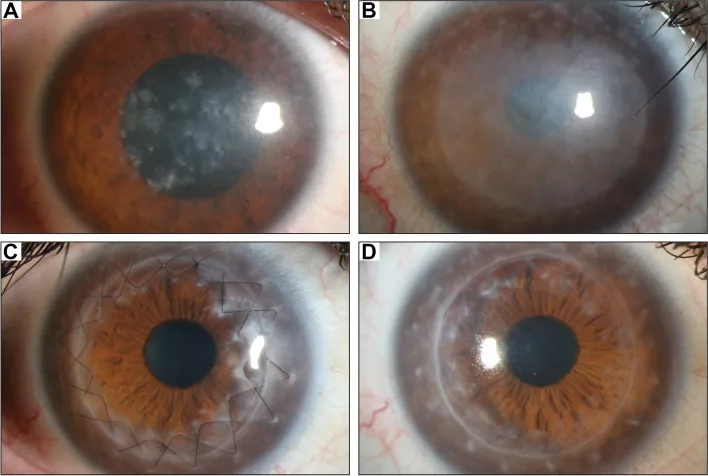
Gassel CJ, et al. Histological findings of corneal tissue after failed phototherapeutic keratectomy in macular corneal dystrophy - a case report. BMC Ophthalmol. 2022. Figure 1. PMCID: PMC9080147. License: CC BY.
Фотография щелевой лампы, показывающая диффузное серовато-белое помутнение и пятнистые отложения от центра до всей роговицы. Это демонстрирует типичные клинические признаки макулярной дистрофии роговицы, облегчая понимание помутнения роговицы, вызывающего снижение зрения.
Типичная картина прогрессирования клинических признаков следующая.
Клинически в строме роговицы наблюдаются диффузные мелкие отложения, и она становится мутной, как матовое стекло. Помутнение прогрессирует, захватывая всю толщину стромы, и распространяется от центра к периферии. Впоследствии, помимо легкого помутнения, появляются многочисленные мелкие серовато-белые помутнения неправильной формы в поверхностных и глубоких слоях стромы.9)
Ранние признаки
Пятнистые помутнения : мелкие серовато-белые пятнистые помутнения появляются в поверхностных слоях стромы в центре роговицы
Помутнение по типу матового стекла : диффузное легкое помутнение стромы роговицы
Нечеткие границы : края помутнения нечеткие, граница с нормальной стромой неясна
Признаки прогрессирующей стадии
Распространение на всю толщину: помутнение затрагивает всю толщу стромы
Расширение к периферии: помутнение распространяется от центра к периферии
Истончение роговицы: толщина центральной роговицы уменьшается
Отложения на эндотелии и десцеметовой мембране: аномальные вещества накапливаются также в глубоких структурах
При щелевой лампе вся роговица диффузно мутная с серовато-белыми неправильными пятнистыми отложениями. На оптическом срезе отложения центрально поверхностные, а периферически глубокие — характерное распределение. Поражения часто появляются концентрическими узорами8).
Помутнение может распространяться на лимб, что является важным отличием MCD от других дистрофий роговицы. При гранулярной и решетчатой дистрофии лимб часто остается прозрачным, тогда как при MCD вся роговица до лимба часто мутная2,8). Неправильный астигматизм связан с отложениями в передней строме, может наблюдаться снижение чувствительности роговицы. Эндотелиальные отложения могут вызывать отек стромы из-за эндотелиальной дисфункции в запущенных случаях8).
Естественное течение варьирует, но часто проходит следующие этапы.
Раннее детство (бессимптомная стадия): генная мутация присутствует с рождения, но данные щелевой лампы скудны, пациент бессимптомен
Школьный возраст — подростковый (стадия раннего помутнения): в передней строме появляется диффузное легкое помутнение, затем наблюдаются пятнистые отложения
10–30 лет (стадия снижения зрения): помутнение прогрессирует, пациент замечает ухудшение зрения
30–40 лет (прогрессирующая стадия): помутнение распространяется на всю строму и лимб, становятся явными снижение чувствительности роговицы, истончение и неправильный астигматизм
Средний и пожилой возраст (стадия показаний к пересадке роговицы): зрительная функция снижена настолько, что мешает повседневной жизни, рассматривается трансплантация
MCD — прогрессирующее заболевание с непрерывным снижением зрительной функции на протяжении всей жизни, что отличает его от гранулярной дистрофии I типа и решетчатой дистрофии (некоторые подтипы), где нарушение зрения остается легким4,8).
Ответственный ген — CHST6 (карбогидрат сульфотрансфераза 6)3). Он расположен на 16q22 и кодирует фермент, переносящий сульфатную группу на N-ацетилглюкозамин в протеогликанах роговицы. Мутации этого гена очень разнообразны: миссенс-, нонсенс-, мутации со сдвигом рамки считывания, делеции в 5’-области, сообщенные в различных этнических группах3,7).
Наследование аутосомно-рецессивное, поэтому оба родителя пробанда обычно являются носителями. Частота заболевания особенно высока в регионах или группах с частыми кровнородственными браками. Компаунд-гетерозиготы, рожденные в браках между разными семьями, также могут заболеть7).
Akama и соавт. в 2000 году идентифицировали CHST6 как ген, ответственный за это заболевание, показав, что как иммунофенотип I, так и II вызываются мутациями одного и того же локуса3). Это открытие показало, что различия в иммунофенотипе определяются разными паттернами мутаций одного гена, что стало основой для последующей генетической диагностики.
Сообщено более 200 мутаций, наиболее частыми являются миссенс-мутации. Sultana и соавт. идентифицировали множество новых мутаций у пациентов из Южной Индии, показав, что высокая частота в этом регионе обусловлена накоплением популяционно-специфических мутаций7). Аналогичные региональные накопления сообщены в Саудовской Аравии и Исландии, что предполагает вклад исторического фона популяции (эффект основателя, обычаи кровнородственных браков) в частоту заболевания5,6,7).
Частота варьирует в зависимости от региона; это относительно редкое заболевание9). По сравнению с другими дистрофиями роговицы, такими как гранулярная дистрофия, случаев меньше, и они чаще сообщаются в семьях с компаунд-гетерозиготностью или кровнородственными браками.
Семейный анамнез: аутосомно-рецессивное наследование, оба родителя должны быть носителями
Кровнородственные браки: повышают заболеваемость
Географический регион: высокая распространенность в Южной Индии, Саудовской Аравии, Исландии, Скандинавии5,7)
QНеобходимо ли генетическое тестирование?
A
Тестирование гена CHST6 полезно для подтверждения диагноза. Оно может быть проведено в аккредитованных медицинских учреждениях. Однако во многих учреждениях основным методом является клиническая диагностика с помощью щелевой лампы. Тестирование полезно для оценки риска у будущих детей или для подтверждения диагноза в случаях с нетипичной клинической картиной.
Подозрение на MCD возникает при наличии триады: двустороннее, прогрессирующее, диффузное помутнение всей роговицы у подростков или молодых взрослых. Сначала подробно собирают анамнез субъективных симптомов (снижение зрения, светобоязнь, чувство раздражения), семейный анамнез и наличие близкородственных браков. Затем проводят оценку роговицы с помощью щелевой лампы, оценку функции эндотелия и, при необходимости, генетическое тестирование.
Это основное исследование для диагностики. При двустороннем помутнении роговицы без гиперемии или отека роговицы следует заподозрить дистрофию роговицы. Для MCD характерны следующие признаки:
Диффузное помутнение по типу матового стекла : вся роговица диффузно мутная
Концентрическое распределение : при срезе щелевой лампой в центре поверхностные, на периферии глубокие
Лимбальная инфильтрация : помутнение может распространяться до лимба
Аномалии эндотелия : в запущенных случаях могут наблюдаться каплевидные отложения
В то время как большинство дистрофий роговицы при исследовании с помощью щелевой лампы выглядят как прерывистые поражения (с прозрачными участками между отложениями), MCD исключительно представляет диффузный паттерн помутнения. Она характеризуется как один из типичных примеров «диффузно наблюдаемых отложений в роговице» наряду с решетчатой дистрофией роговицы I типа и желатинозной каплевидной дистрофией роговицы.
Оптическая когерентная томография переднего сегмента (AS-OCT) : визуализирует распределение отложений в поверхностных и глубоких слоях роговицы
Конфокальная микроскопия in vivo : показывает высокорефлективный материал с нечеткими границами и исчезновение нормальных кератоцитов8)
Топография роговицы : показывает повышенную плотность в области вершины роговицы и истончение центральной роговицы
Ультразвуковая биомикроскопия (УБМ) : полезна для оценки глубоких помутнений и задней структуры роговицы
Зеркальный микроскоп : оценка плотности и морфологии эндотелиальных клеток. Степень поражения эндотелия напрямую влияет на выбор хирургической техники
Роговичный Pentacam (устройство для съемки по Шаймпфлюгу) : позволяет получить карту плотности всей толщи роговицы, полезна для трехмерной оценки участков помутнения
Гистологически окрашивание альциановым синим и коллоидным железом положительно, и наблюдается диффузное накопление гипосульфатированных гликозаминогликанов внутри и снаружи кератоцитов стромы роговицы2,8). Могут наблюдаться разрывы мембраны Боумена, а в запущенных случаях аномальные вещества также обнаруживаются в эндотелиальных клетках. На мембране Десцемета могут также наблюдаться изменения, напоминающие капли (guttae).
Четко очерченные гранулярные отложения (с прозрачными участками между ними)
Решетчатая дистрофия роговицы I типа
Аутосомно-доминантный
Линейные и сетчатые решетчатые линии (амилоид)
Желатинозная каплевидная дистрофия роговицы
Аутосомно-рецессивный
Возвышающиеся поражения в виде тутовой ягоды или полос
Кристаллическая дистрофия роговицы Шнайдера
Аутосомно-доминантный
Игольчатые кристаллы, ассоциированные с гиперлипидемией
Врожденная стромальная дистрофия роговицы
Аутосомно-доминантный
Присутствует с рождения, утолщение стромы
Системный мукополисахаридоз (роговичный тип)
Рецессивный / X-сцепленный
С системными симптомами
Кроме того, в дифференциальный диагноз включаются задняя полиморфная дистрофия роговицы (PPCD) и преддесцеметовая дистрофия роговицы (PDCD). Системные мукополисахаридозы (синдромы Гурлера, Шейе, Моркио и др.) также могут вызывать помутнение роговицы, поэтому необходима оценка, включающая системные проявления8).
Клинический диагноз ставится на основании щелевой лампы, выявляющей диффузное помутнение роговицы по типу матового стекла и пятнистые отложения, двусторонний прогрессирующий характер, семейный анамнез и возраст начала (10–30 лет). Для подтверждения полезно генетическое тестирование гена CHST6.
Цели лечения MCD: (1) сохранение/восстановление зрительной функции, (2) облегчение боли и симптомов раздражения путем стабилизации поверхности глаза, (3) предотвращение осложнений (эрозия эпителия, инфекция). Поскольку этиотропного лечения не существует, проводится поэтапное вмешательство в зависимости от стадии и симптомов.
При прогрессирующем снижении зрения трансплантация роговицы является единственным радикальным методом лечения. Метод операции выбирается в зависимости от наличия или отсутствия поражения эндотелия.
Глубокая передняя послойная кератопластика (DALK) : метод первого выбора при отсутствии поражения эндотелия8,9). Сохраняя собственный эндотелий роговицы пациента, риск отторжения трансплантата ниже, чем при сквозной кератопластике. В оценочном отчете AAO (Американской академии офтальмологии) DALK оценивается как обеспечивающая восстановление зрительной функции, эквивалентное PKP, при меньшей потере эндотелия8). Частота рецидивов после DALK низкая, и замена стромы роговицы реципиента делает рецидив маловероятным.
Сквозная кератопластика (PKP) : показана в запущенных случаях с отложениями аномального вещества в эндотелии или мембране Десцемета, а также при выраженном истончении центральной роговицы7). Средний возраст первой PKP при MCD составляет 30–40 лет7), выживаемость трансплантата хорошая.
Фототерапевтическая кератэктомия (PTK) : симптоматическое лечение рецидивирующих эрозий эпителия роговицы и поверхностных рубцовых помутнений. Однако следует учитывать индукцию гиперметропии и стромального помутнения.
Поскольку при MCD аномальные вещества могут откладываться и в эндотелиальных клетках, при глубоком распространении заболевания PKP имеет тенденцию предпочитаться DALK7). При эндотелиальных аномалиях показана сквозная кератопластика. Детальная предоперационная оценка эндотелия (зеркальная, конфокальная микроскопия) является ключевой для определения метода операции.
Частота рецидивов после DALK низкая, составляет около 5%. Выживаемость трансплантата после PKP хорошая, во многих сообщениях отмечается долгосрочное выживание, хотя также описаны случаи повторного отложения аномального вещества на трансплантате через несколько лет или десятилетий после операции 8). В саудовской серии Al-Swailem и соавт. выживаемость после PKP при MCD была хорошей, но в некоторых случаях наблюдался рецидив при длительном наблюдении 8). Кроме того, в обзоре AAO Reinhart и соавт. было показано, что DALK при стромальных дистрофиях обеспечивает равное или лучшее восстановление зрения и низкую скорость потери эндотелия по сравнению с PKP9). Многоцентровое исследование Unal и соавт. также сообщило об эффективности DALK при стромальных дистрофиях, включая MCD.
В целом, послеоперационный рецидив трансплантатов роговицы возникает из-за того, что молекулярный патогенез основного заболевания сохраняется в организме реципиента. При DALK сохраняется эндотелий и преддесцеметовый слой реципиента, поэтому следует учитывать, что заболевание может прогрессировать в случаях с поражением эндотелия. После операции необходимо длительное регулярное наблюдение (острота зрения, щелевая лампа, измерение плотности эндотелиальных клеток).
QЧто выбрать: DALK или PKP?
A
Если эндотелий или десцеметова мембрана не поражены, методом выбора является глубокая передняя послойная кератопластика (DALK). DALK сохраняет собственный эндотелий роговицы, что снижает риск отторжения, а частота послеоперационных рецидивов составляет около 5%. С другой стороны, в случаях, когда аномальные отложения затрагивают и эндотелий, показана сквозная кератопластика (PKP). Решение о методе операции принимается на основе предоперационной оценки эндотелия.
Ген CHST6 кодирует углеводную сульфотрансферазу 6 3). Этот фермент отвечает за перенос сульфатной группы на N-ацетилглюкозамин в молекуле кератана и необходим для нормального синтеза кератансульфата (KS), входящего в состав протеогликанов роговицы.
Потеря активности фермента из-за мутации гена приводит к синтезу гипосульфатированного кератансульфата с недостаточным сульфатированием. Этот аномальный кератансульфат обладает низкой растворимостью и аномально откладывается внутри и снаружи кератоцитов стромы роговицы 2,3).
Количественные и качественные аномалии кератансульфата приводят к следующему каскаду патологических изменений.
Аномальная продукция малых протеогликанов (SLRP): Специфичные для роговицы SLRP, такие как люмикан, кератокан и мимекан, не синтезируются нормально.
Аномальное расположение коллагеновых волокон: Эти SLRP строго контролируют диаметр и межволоконное расстояние коллагеновых волокон роговицы, обеспечивая прозрачность. Снижение функции SLRP приводит к неоднородности диаметра коллагеновых волокон и изменению межволоконного расстояния2).
Аномальное накопление во внеклеточном матриксе: Несульфатированный кератан сам по себе откладывается во внеклеточном матриксе.
Увеличение рассеяния света и потеря прозрачности: Вышеуказанные комбинированные изменения усиливают рассеяние видимого света, и вся роговица становится диффузно мутной.
Накопление гликозаминогликанов наблюдается внутри и снаружи стромальных кератоцитов, и по мере прогрессирования поражения оно распространяется на мембрану Боумена, мембрану Десцемета и эндотелиальные клетки. При I типе снижение ферментативной активности также подтверждено в ушном хряще, что позволяет предположить, что это может быть частичным проявлением системного нарушения метаболизма кератансульфата2). Однако клинически системные симптомы редки, и заболевание рассматривается как локализованное с преимущественным поражением роговицы.
Среди протеогликанов роговицы люмикан контролирует диаметр коллагеновых волокон примерно до 25 нм, в то время как кератокан и мимекан поддерживают равномерное межволоконное расстояние. Когда сульфатированные боковые цепи этих SLRP короткие и неполные, коллагеновые волокна имеют вариабельность диаметра, а межволоконное расстояние становится неравномерным. В результате увеличивается рассеяние света в строме роговицы, что клинически наблюдается как помутнение по типу матового стекла.
Для поддержания прозрачности роговицы классически известна решетчатая теория Маурера, основанная на «гашении интерференции света упорядоченным решетчатым расположением коллагеновых волокон». При MCD эта решетчатая структура нарушается из-за аномалии SLRP, что приводит к потере прозрачности2,4).
Недавние фундаментальные исследования сообщили, что дисфункция аутофагии, вызванная мутацией CHST6, может индуцировать пироптоз (воспалительную гибель клеток) кератоцитов, способствуя прогрессированию заболевания8). Подобные аномалии аутофагии были зарегистрированы и при других дистрофиях роговицы (например, гранулярной II типа), и они привлекают внимание как общая патология всех дистрофий роговицы.
Генная таргетная терапия предлагается в качестве стратегии постоянного лечения 8). Фундаментальные исследования редактирования генов с использованием CRISPR/Cas9 продвигаются при дистрофии эпителия роговицы Меесмана, и в будущем они могут стать вариантом лечения и для MCD. Однако для клинического применения остается множество проблем, включая непреднамеренное редактирование нормальных аллелей (внецелевые эффекты), создание эффективных методов переноса генов в кератоциты стромы роговицы и проверку долгосрочной безопасности.
Фундаментально изучается подход к ферментативному удалению накопленного гипосульфатированного кератансульфата в роговице. Это локальное применение концепции ферментозаместительной терапии, которая уже используется при системных мукополисахаридозах, но в настоящее время сообщений о клиническом применении нет.
Эндотелиальная кератопластика с отслоением десцеметовой мембраны (DSAEK) и эндотелиальная кератопластика десцеметовой мембраны (DMEK) - это методы, разработанные в основном для эндотелиальной дистрофии Фукса и буллезной кератопатии, но их применение при MCD с преимущественным поражением эндотелия является предметом будущих исследований. В настоящее время сквозная кератопластика (PKP) является реалистичным выбором для этого заболевания, поражающего как строму, так и эндотелий. В будущем была предложена концепция «последовательной кератопластики», сочетающей DALK (глубокую переднюю послойную кератопластику) для замены только стромы и DMEK для замены только эндотелия, но все это находится на стадии исследований.
Уровень кератансульфата в сыворотке крови, как сообщается, ниже нормы у пациентов с MCD иммунофенотипа II, и обсуждается его полезность в качестве системного метаболического маркера 2). В будущем ожидается его применение для скрининга с помощью анализа крови и ранней диагностики носителей в семьях с мутациями CHST6. Кроме того, автоматический анализ изображений щелевой лампы с помощью искусственного интеллекта является областью исследований, которая может способствовать раннему выявлению редких заболеваний, таких как это.
Поскольку это аутосомно-рецессивное заболевание, примерно 25% сибсов пациента могут быть поражены. Использование генетического тестирования CHST6 позволяет проводить семейный скрининг и генетическое консультирование по вопросам брака и беременности. Раннее направление к специалисту рекомендуется в семьях с историей кровного родства или когда у членов семьи наблюдаются аналогичные помутнения роговицы.
Akama TO, Nishida K, Nakayama J, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nature Genetics. 2000;26(2):237-241. doi:10.1038/79987.
Aggarwal S, Peck T, Golen J, Karcioglu ZA. Macular corneal dystrophy: A review. Survey of Ophthalmology. 2018;63(5):609-617. doi:10.1016/j.survophthal.2018.03.004.
Jonasson F, Oshima E, Thonar EJ, et al. Macular corneal dystrophy in Iceland. A clinical, genealogic, and immunohistochemical study of 28 patients. Ophthalmology. 1996;103(7):1111-1117. doi:10.1016/s0161-6420(96)30559-9.
Jonasson F, Johannsson JH, Garner A, Rice NS. Macular corneal dystrophy in Iceland. Eye (London, England). 1989;3 ( Pt 4):446-54. doi:10.1038/eye.1989.66. PMID:2606219.
Sultana A, Sridhar MS, Jagannathan A, et al. Novel mutations of the carbohydrate sulfotransferase-6 (CHST6) gene causing macular corneal dystrophy in India. Mol Vis. 2003;9:730-734. PMID:14735064.
Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating keratoplasty for macular corneal dystrophy. Ophthalmology. 2005;112(2):220-224. doi:10.1016/j.ophtha.2004.08.017.
Reinhart WJ, Musch DC, Jacobs DS, Lee WB, Kaufman SC, Shtein RM. Deep anterior lamellar keratoplasty as an alternative to penetrating keratoplasty a report by the american academy of ophthalmology. Ophthalmology. 2011;118(1):209-218. doi:10.1016/j.ophtha.2010.11.002. PMID:21199711.
Скопируйте текст статьи и вставьте его в выбранный ИИ-ассистент.
Статья скопирована в буфер обмена
Откройте ИИ-ассистент ниже и вставьте скопированный текст в чат.