دیستروفی ماکولار قرنیه (macular corneal dystrophy: MCD) یک دیستروفی قرنیه ارثی است که در آن گلیکوزآمینوگلیکانها (عمدتاً کراتان سولفات) در استرومای قرنیه تجمع مییابند. این بیماری به صورت اتوزومال مغلوب به ارث میرسد و علت آن جهش در ژن CHST6 واقع در بازوی بلند کروموزوم ۱۶ (16q22) است1,3). در گذشته به آن دیستروفی قرنیه گرنو نوع II یا دیستروفی قرنیه فِر نیز میگفتند.
در حالی که بسیاری از دیستروفیهای استرومای قرنیه دیگر (گرانولار و لاتیس) به صورت اتوزومال غالب به ارث میرسند، ویژگی بارز این بیماری وراثت اتوزومال مغلوب است. در ژاپن، این بیماری همراه با دیستروفی گرانولار قرنیه (نوع I و II)، دیستروفی لاتیس قرنیه (نوع I و IIIA)، و دیستروفی ژلاتینوز دراپ-لایک قرنیه به عنوان چهار دیستروفی بزرگ قرنیه شناخته میشود و این چهار بیماری حدود ۹۶٪ از کل دیستروفیهای قرنیه را تشکیل میدهند. از این میان، دو نوع اول (گرانولار و لاتیس) اتوزومال غالب و دو نوع آخر (ژلاتینوز دراپ-لایک و ماکولار) اتوزومال مغلوب هستند.
بر اساس طبقهبندی IC3D (کمیته بینالمللی طبقهبندی دیستروفیهای قرنیه)، MCD به عنوان یکی از انواع دیستروفی استروما (لایه میانی قرنیه) در نظر گرفته میشود 1). در مقاله مروری Aggarwal و همکاران در مجله Survey of Ophthalmology سال 2018، این بیماری به عنوان «دیستروفی استروما نادر اما با تأثیر قابل توجه بر عملکرد بینایی» شرح داده شده و سیستم تشخیص و درمان آن خلاصه شده است 4). در ویرایش دوم طبقهبندی IC3D، دیستروفیهای قرنیه به دستههای 1 تا 4 تقسیم میشوند که بر اساس قدرت شواهد ژنتیکی، یافتههای پاتولوژیک و تصویر بالینی طبقهبندی میگردند 1). MCD با شناسایی جهش در ژن CHST6 در دسته 1 (دیستروفی تأیید شده در سطح ژن) قرار میگیرد.
از نظر پیشینه تاریخی، اولین بار در سال 1890 توسط Groenouw توصیف شد و بعداً دیستروفی دانهای (گرانولار) به عنوان «نوع I» و دیستروفی ماکولار (لکهای) به عنوان «نوع II» نامیده شد. در سال 1938، Jones و Zimmerman آن را به عنوان یک بیماری مستقل معرفی کردند و در سال 2000، Akama و همکاران با شناسایی ژن CHST6، اساس مولکولی آن را روشن ساختند 3).
در سطح جهانی، تفاوتهای منطقهای زیادی وجود دارد و در مناطق با شیوع بالا، تجمع خانوادگی مشاهده میشود. این بیماری نسبتاً نادر است.
تفاوتهای منطقهای در شیوع
ایالات متحده: حدود 0.3 نفر در 250,000 نفر، نادر است 2,3)
ایسلند: حدود 19 نفر در 250,000 نفر، یکی از مناطق با بالاترین شیوع در جهان 5,6)
مناطق با شیوع بالا: جنوب هند، عربستان سعودی، ایسلند و کشورهای اسکاندیناوی 5,7)
سایر مناطق: نسبتاً نادر. در ازدواجهای فامیلی و هتروزیگوتهای مرکب بروز میکند
فنوتیپ ایمنی
نوع I: کراتان سولفات در قرنیه و سرم منفی است 2)
نوع IA: در کراتوسیتهای قرنیه مثبت، در سرم منفی است 2)
نوع II: کراتان سولفات در قرنیه و سرم مثبت است 2)
تصویر بالینی: هر سه نوع فنوتیپ یکسان دارند و با لامپ شکاف (اسلیت لمپ) قابل تفکیک نیستند 2,8)
فنوتیپ ایمنی MCD بر اساس میزان کراتان سولفات در قرنیه و سرم با استفاده از آنتیبادی مونوکلونال ضد کراتان سولفات طبقهبندی میشود 2,3).
فنوتیپ
کراتان سولفات قرنیه
کراتان سولفات سرم
نوع I
منفی
منفی
نوع IA
مثبت (داخل سلولی)
منفی
نوع II
مثبت
مثبت
بیشتر بیماران به نوع I یا IA طبقهبندی میشوند. با این حال، از نظر بالینی تمایز این زیرگروهها مهم نیست و با معاینه قابل تشخیص نیستند2,8).
Qدیستروفی ماکولار قرنیه چه تفاوتی با سایر دیستروفیهای قرنیه دارد؟
A
بزرگترین تفاوت در توارث اتوزومال مغلوب است. دیستروفی گرانولار و لاتیس قرنیه توارث اتوزومال غالب دارند، اما این بیماری نیاز به جهش در هر دو آلل ژن CHST6 دارد. همچنین کدورت منتشر شیشهای (ground-glass) ایجاد میکند و کل قرنیه کدر میشود. در معاینه با لامپ شکاف، رسوبات در مرکز در لایههای سطحی و در محیط در لایههای عمقی دیده میشود.
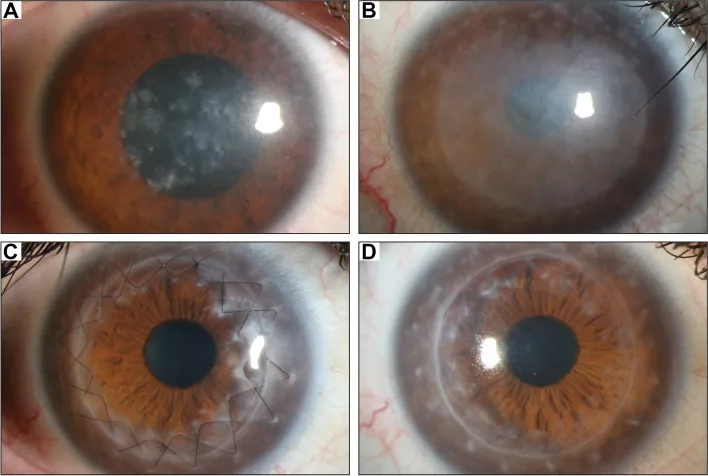
Gassel CJ, et al. Histological findings of corneal tissue after failed phototherapeutic keratectomy in macular corneal dystrophy - a case report. BMC Ophthalmol. 2022. Figure 1. PMCID: PMC9080147. License: CC BY.
در عکس با میکروسکوپ شکافدار، کدورت منتشر خاکستری-سفید و رسوبات لکهای از مرکز تا کل قرنیه دیده میشود. این تصویر یافتههای بالینی مشخصه دیستروفی ماکولار قرنیه را نشان میدهد و به درک کدورت قرنیه که باعث کاهش بینایی میشود کمک میکند.
الگوی پیشرفت معمول یافتههای بالینی به شرح زیر است:
از نظر بالینی، رسوبات ریز به صورت منتشر در استرومای قرنیه دیده میشود و قرنیه ظاهری شیشهمات پیدا میکند. با پیشرفت، کدورت به تمام ضخامت استروما گسترش مییابد و از مرکز به سمت محیط پخش میشود. سپس، علاوه بر کدورت خفیف، کدورتهای متعدد کوچک و نامنظم خاکستری-سفید در لایههای سطحی تا عمقی استروما مشاهده میشود.9)
یافتههای اولیه
کدورتهای لکهای: کدورتهای کوچک خاکستری-سفید در لایه سطحی استرومای مرکزی قرنیه ظاهر میشوند
کدورت شیشهمات: کدورت منتشر خفیف در استرومای قرنیه مشاهده میشود
مرز نامشخص: لبههای کدورت نامشخص است و مرز با استرومای طبیعی به خوبی مشخص نیست
یافتههای مرحله پیشرفته
گسترش به تمام لایهها: کدورت به تمام ضخامت استروما میرسد
گسترش به نواحی محیطی: کدورت از مرکز به سمت محیط گسترش مییابد
نازک شدن قرنیه: ضخامت قرنیه مرکزی کاهش مییابد
رسوب در اندوتلیوم و غشای دسمه: مواد غیرطبیعی در ساختارهای عمقی نیز تجمع مییابند
در معاینه با لامپ شکاف، کل قرنیه به طور منتشر کدر است و رسوبات نامنظم خاکستری-سفید در آن دیده میشود. با ایجاد برش نوری با نور شکاف، توزیع مشخصهای نشان میدهد: در ناحیه مرکزی، رسوبات در لایههای سطحی و در ناحیه محیطی، در لایههای عمقی قرار دارند. ضایعات لکهای اغلب به صورت دایرههای متحدالمرکز ظاهر میشوند8).
کدورت ممکن است به لیمبوس نیز گسترش یابد که این نکته یک تمایز مهم از سایر دیستروفیهای قرنیه است. در دیستروفی قرنیه گرانولار و دیستروفی قرنیه لتیس، لیمبوس اغلب شفاف باقی میماند، در حالی که در MCD، کل قرنیه تا لیمبوس اغلب کدر میشود2,8). همچنین، ظهور آستیگماتیسم نامنظم با رسوبات استرومای قدامی مرتبط است و ممکن است کاهش حس قرنیه نیز مشاهده شود. از آنجایی که مواد غیرطبیعی در اندوتلیوم نیز رسوب میکنند، در موارد پیشرفته ممکن است ادم استرومایی ناشی از کاهش عملکرد اندوتلیوم رخ دهد8).
سیر طبیعی بین افراد متفاوت است، اما اغلب مراحل زیر را طی میکند.
دوران کودکی (مرحله بدون علامت): جهش ژنتیکی از بدو تولد وجود دارد، اما یافتههای لامپ شکاف اندک و بدون علامت است
سن مدرسه تا بلوغ (مرحله کدورت اولیه): کدورت منتشر خفیف در لایههای سطحی استرومای قرنیه ظاهر میشود و به تدریج رسوبات لکهای مشاهده میشود
سن ۱۰ تا ۳۰ سال (مرحله کاهش بینایی): کدورت پیشرفت میکند و بیمار متوجه کاهش بینایی میشود
دهه ۳۰ تا ۴۰ (مرحله پیشرفته): کدورت به تمام ضخامت استروما و لیمبوس گسترش مییابد و کاهش حس قرنیه، نازک شدن قرنیه و آستیگماتیسم نامنظم آشکار میشود
میانسالی تا سالمندی (مرحله مناسب برای پیوند قرنیه): کاهش عملکرد بینایی به سطحی میرسد که در فعالیتهای روزمره اختلال ایجاد میکند و پیوند قرنیه در نظر گرفته میشود
MCD یک بیماری پیشرونده است و کاهش عملکرد بینایی در طول زندگی ادامه مییابد، که از این نظر پیشآگهی آن با دیستروفی قرنیه گرانولار نوع I و برخی زیرگروههای دیستروفی قرنیه لتیس که اختلال بینایی در آنها خفیف است، تفاوت زیادی دارد4,8).
ژن عامل بیماری CHST6 (کربوهیدرات سولفوترانسفراز 6) است3). این ژن در موقعیت 16q22 قرار دارد و آنزیمی را کد میکند که گروه سولفات را به N-استیلگلوکزآمین روی پروتئوگلیکانهای قرنیه منتقل میکند. انواع جهشهای این ژن بسیار زیاد است و جهشهای missense، nonsense، frameshift و حذف در ناحیه بالادست 5’ در اقوام مختلف گزارش شده است3,7).
از آنجا که این بیماری با وراثت اتوزومال مغلوب منتقل میشود، معمولاً هر دو والدین فرد مبتلا ناقل هستند. شیوع بیماری به ویژه در مناطق و جمعیتهایی با ازدواج فامیلی بالا بیشتر است. همچنین در افراد با هتروزیگوت مرکب ناشی از ازدواج بین دو خانواده مختلف نیز ممکن است بیماری بروز کند7).
Akama و همکاران در سال 2000 ژن CHST6 را به عنوان ژن عامل این بیماری شناسایی کردند و نشان دادند که هر دو فنوتیپ ایمنی نوع I و II ناشی از جهش در همان جایگاه ژنی هستند3). این کشف یک یافته مهم است که نشان میدهد تفاوت در فنوتیپ ایمنی توسط الگوهای مختلف جهش در یک ژن واحد تعیین میشود و پایهای برای سیستمهای تشخیص ژنتیکی بعدی شد.
بیش از 200 نوع جهش گزارش شده است که شایعترین آنها جهشهای missense هستند. Sultana و همکاران جهشهای جدید متعددی را در گروهی از بیماران هندی جنوبی شناسایی کردند و نشان دادند که فراوانی بالای این بیماری در آن منطقه ناشی از تجمع جهشهای خاص منطقهای است7). تجمع مشابه منطقهای در عربستان سعودی و ایسلند نیز گزارش شده است و به نظر میرسد پیشینه تاریخی جمعیت (اثر بنیانگذار و عادات ازدواج فامیلی) در شیوع بیماری نقش دارد5,6,7).
شیوع بیماری بسته به منطقه متفاوت است و این بیماری نسبتاً نادر است9). در مقایسه با سایر دیستروفیهای قرنیه مانند دیستروفی قرنیه دانهای، موارد ابتلا کمتر است و معمولاً در خانوادههایی با سابقه ازدواج فامیلی یا هتروزیگوت مرکب گزارش میشود.
سابقه خانوادگی: به دلیل وراثت اتوزومال مغلوب، هر دو والدین باید ناقل باشند
ازدواج فامیلی: احتمال بروز بیماری را افزایش میدهد
منطقه جغرافیایی: شیوع در جنوب هند، عربستان سعودی، ایسلند و کشورهای اسکاندیناوی بالاتر است5,7)
Qآیا آزمایش ژنتیک ضروری است؟
A
آزمایش ژن CHST6 برای تشخیص قطعی مفید است. آزمایش ژنتیک در مراکز درمانی معتبر قابل انجام است. با این حال، در بسیاری از مراکز، تشخیص بالینی با میکروسکوپ اسلیت لامپ اصلیترین روش است. این آزمایش برای ارزیابی خطر ابتلا در فرزندان آینده و تشخیص قطعی در مواردی که یافتههای بالینی معمولی نیستند، مفید است.
نشانهای که باعث شک به MCD میشود «دوچشمی، پیشرونده، کدورت منتشر کل قرنیه در نوجوانان تا بزرگسالان جوان» است. ابتدا سابقه علائم ذهنی (کاهش بینایی، فوتوفوبی، احساس تحریک)، سابقه خانوادگی و ازدواج فامیلی به طور دقیق گرفته میشود. سپس ارزیابی یافتههای قرنیه با میکروسکوپ اسلیت لمپ، ارزیابی عملکرد اندوتلیوم و در صورت لزوم آزمایش ژنتیک انجام میشود.
این آزمایش پایهای تشخیص است. در صورت مشاهده کدورت قرنیه دوچشمی بدون پرخونی یا ادم قرنیه، باید به دیستروفی قرنیه مشکوک شد. در MCD یافتههای زیر مشخصه هستند:
کدورت منتشر شیشهای : کل قرنیه به طور منتشر کدر میشود
رسوبات لکهای : کدورتهای متعدد نامنظم به رنگ سفید مایل به خاکستری
توزیع هممرکز : با برش نور اسلیت، در مرکز لایههای سطحی و در محیط لایههای عمقی دیده میشود
درگیری لیمبوس : کدورت ممکن است تا لیمبوس گسترش یابد
ناهنجاری سطح اندوتلیوم : در موارد پیشرفته ممکن است رسوبات قطرهای شکل دیده شود
در حالی که بسیاری از دیستروفیهای قرنیه در سطح اسلیت به صورت ضایعات ناپیوسته (با نواحی شفاف بین رسوبات) مشاهده میشوند، MCD به طور استثنایی الگوی کدورت منتشر را نشان میدهد. همراه با دیستروفی قرنیه مشبک نوع I و دیستروفی قرنیه ژلاتینی قطرهای، به عنوان نمونهای از «رسوبات قرنیهای که به صورت منتشر مشاهده میشوند» توصیف میشود.
از نظر بافتشناسی، رنگآمیزی آلسین بلو و رنگآمیزی کلوئیدی آهن مثبت بوده و تجمع منتشر گلیکوزآمینوگلیکانهای کم سولفات در داخل و خارج سلولهای کراتوسیت استرومای قرنیه مشاهده میشود2,8). ممکن است پارگی غشای بومن دیده شود و در موارد پیشرفته، مواد غیرطبیعی در داخل سلولهای اندوتلیال نیز یافت میشود. گاهی اوقات یافتههای شبیه گوتات (guttae) روی غشای دسمه مشاهده میشود.
رسوبات دانهای با مرز مشخص (با نواحی شفاف بین آنها)
دیستروفی شبکهای قرنیه نوع I
اتوزومال غالب
خطوط شبکهای خطی و مشبک (آمیلوئید)
دیستروفی قرنیه ژلاتینی قطرهای
اتوزومال مغلوب
ضایعات برجسته توتمانند و نواری
دیستروفی قرنیه کریستالی اشنیدر
اتوزومال غالب
کریستالهای سوزنی شکل همراه با هیپرلیپیدمی
دیستروفی مادرزادی استرومای قرنیه
اتوزومال غالب
وجود از بدو تولد و ضخیم شدن استروما
موکوپلیساکاریدوز سیستمیک (نوع قرنیهای)
مغلوب / وابسته به X
همراه با علائم سیستمیک
علاوه بر این، دیستروفی قرنیه خلفی پلیمورف (PACD) و دیستروفی قرنیه پیشدسامه (PDCD) نیز در تشخیص افتراقی مطرح میشوند. موکوپلیساکاریدوز سیستمیک (مانند سندرمهای هورلر، شای، مورکیو) نیز میتواند باعث کدورت قرنیه شود، بنابراین ارزیابی همراه با یافتههای سیستمیک ضروری است8).
Qدیستروفی قرنیه ماکولار چگونه تشخیص داده میشود؟
A
با استفاده از لامپ شکافی، کدورت منتشر شیشهمات قرنیه و رسوبات ماکولار تأیید میشود و با توجه به دوطرفه و پیشرونده بودن، سابقه خانوادگی و سن شروع (۱۰ تا ۳۰ سال) تشخیص بالینی داده میشود. برای تشخیص قطعی، آزمایش ژنتیکی CHST6 مفید است.
اهداف درمان MCD عبارتند از: (1) حفظ و بازیابی عملکرد بینایی، (2) کاهش درد و علائم تحریکی با تثبیت سطح چشم، و (3) پیشگیری از عوارض (فرسایش اپیتلیال و عفونت). از آنجایی که درمان علتشناختی وجود ندارد، مداخلات مرحلهای بر اساس مرحله بیماری و علائم انجام میشود.
در مواردی که کاهش بینایی پیشرفت کرده است، پیوند قرنیه تنها درمان قطعی است. روش جراحی بر اساس وجود یا عدم وجود درگیری اندوتلیال انتخاب میشود.
کراتوپلاستی لایهای عمیق قدامی (DALK): در مواردی که اندوتلیال درگیر نشده است، انتخاب اول است8,9). با حفظ اندوتلیال خود بیمار، خطر رد پیوند کمتر از پیوند تماملایه است. در گزارش ارزیابی آکادمی چشمپزشکی آمریکا (AAO)، DALK برای دیستروفیهای استرومایی بهبود بینایی مشابه PKP با از دست دادن کمتر اندوتلیال ارزیابی شده است8). میزان عود پس از DALK پایین گزارش شده است و به دلیل جایگزینی استرومای گیرنده، عود نادر است.
کراتوپلاستی نافذ (PKP): در موارد پیشرفته با رسوب مواد غیرطبیعی در اندوتلیال و غشای دسمه، و همچنین در موارد نازکشدگی شدید قرنیه مرکزی اندیکاسیون دارد7). میانگین سن اولین PKP برای MCD 30-40 سال گزارش شده است7) و میزان بقای پیوند خوب است.
کراتکتومی درمانی فوتوتراپی (PTK): بهصورت علامتی برای فرسایش مکرر اپیتلیال و کدورتهای اسکار سطحی انجام میشود. با این حال، باید به دوربینی شدن و کدورت استروما توجه داشت.
از آنجایی که در MCD مواد غیرطبیعی ممکن است در سلولهای اندوتلیال نیز رسوب کنند، در مواردی که بیماری به عمق گسترش یافته است، PKP نسبت به DALK ترجیح داده میشود7). در صورت همراهی با ناهنجاری اندوتلیال، پیوند تماملایه اندیکاسیون دارد. ارزیابی دقیق اندوتلیال قبل از عمل (میکروسکوپ اسپکولار و کانفوکال) برای تعیین روش جراحی ضروری است.
نرخ عود پس از DALK پایین گزارش شده است و حدود ۵٪ تخمین زده میشود. میزان بقای پیوند پس از PKP خوب است و در بسیاری از گزارشها بقای طولانیمدت حاصل میشود، اما مواردی از رسوب مجدد مواد غیرطبیعی بر روی پیوند در طی چندین سال تا دههها پس از جراحی نیز گزارش شده است8). در سری مطالعات Al-Swailem و همکاران در عربستان سعودی، میزان بقای پیوند پس از PKP برای MCD خوب بود، اما در برخی موارد عود در طول دوره طولانی مدت مشاهده شد8). همچنین، در مرور AAO توسط Reinhart و همکاران نشان داده شده است که DALK برای دیستروفیهای استرومایی، بازیابی بینایی برابر یا بهتر از PKP و نرخ پایینتر از دست دادن سلولهای اندوتلیال را فراهم میکند9). مطالعه چندمرکزی Unal و همکاران نیز اثربخشی DALK را برای دیستروفیهای استرومایی از جمله MCD گزارش کرده است.
به طور کلی، عود پس از پیوند قرنیه به دلیل باقی ماندن پاتوفیزیولوژی مولکولی بیماری در میزبان رخ میدهد. در DALK، اندوتلیوم و لایه پیش از غشای دسمه میزبان حفظ میشود، بنابراین در مواردی که بیماری اندوتلیال وجود دارد، باید توجه داشت که بیماری ممکن است پیشرفت کند. پس از جراحی، پیگیری منظم طولانیمدت (اندازهگیری بینایی، معاینه با لامپ شکاف، اندازهگیری تراکم سلولهای اندوتلیال) ضروری است.
Qکدام یک را باید انتخاب کرد: DALK یا PKP؟
A
اگر بیماری به اندوتلیوم و غشای دسمه گسترش نیافته باشد، پیوند عمقی لایهای قدامی (DALK) انتخاب اول است. DALKاندوتلیوم قرنیه خود بیمار را حفظ میکند، بنابراین خطر رد پیوند کمتر است و نرخ عود پس از عمل حدود ۵٪ گزارش شده است. از سوی دیگر، در مواردی که رسوب مواد غیرطبیعی به اندوتلیوم نیز گسترش یافته است، پیوند تماملایه (PKP) اندیکاسیون دارد. روش جراحی بر اساس ارزیابی قبل از عمل اندوتلیوم تعیین میشود.
ژن CHST6 کدکننده کربوهیدرات سولفوترانسفراز ۶ است3). این آنزیم مسئول انتقال گروه سولفات به N-استیلگلوکزامین روی مولکول کراتان است و برای سنتز طبیعی کراتان سولفات (KS) موجود در پروتئوگلیکانهای قرنیه ضروری است.
از دست رفتن فعالیت آنزیم در اثر جهش ژنی منجر به سنتز کراتان سولفات با سولفاته ناقص (هیپوسولفاته) میشود. این کراتان سولفات غیرطبیعی حلالیت کمی دارد و در داخل و خارج سلولهای کراتوسیت استرومای قرنیه رسوب میکند2,3).
ناهنجاریهای کمی و کیفی کراتان سولفات منجر به زنجیرهای از تغییرات پاتولوژیک زیر میشود.
تولید غیرطبیعی پروتئوگلیکانهای کوچک غنی از لوسین (SLRP): SLRPهای اختصاصی قرنیه مانند لومیکان، کراتوکان و میمکان به طور طبیعی سنتز نمیشوند.
ناهنجاری در آرایش فیبرهای کلاژن: این SLRPها قطر و فاصله بین فیبرهای کلاژن قرنیه را به دقت تنظیم کرده و شفافیت را تضمین میکنند. کاهش عملکرد SLRP باعث ناهمگنی در قطر فیبرهای کلاژن و تغییر فاصله بین آنها میشود2).
تجمع غیرطبیعی در ماتریکس خارج سلولی: کراتان سولفات غیرسولفاته خود در ماتریکس خارج سلولی رسوب میکند.
افزایش پراکندگی نور و از دست دادن شفافیت: تغییرات ترکیبی فوق باعث افزایش پراکندگی نور مرئی و کدورت منتشر کل قرنیه میشود.
تجمع گلیکوزآمینوگلیکانها در داخل و خارج سلولهای کراتوسیت استروما مشاهده میشود و با پیشرفت بیماری به غشای بومن، غشای دسمه و سلولهای اندوتلیال گسترش مییابد. در نوع I، کاهش فعالیت آنزیمی در غضروف گوش نیز تأیید شده است که نشاندهنده احتمال وجود یک اختلال سیستمیک در متابولیسم کراتان سولفات است2). با این حال، تظاهرات بالینی سیستمیک نادر است و این بیماری به عنوان یک اختلال موضعی با تظاهر اصلی قرنیه در نظر گرفته میشود.
در میان پروتئوگلیکانهای قرنیه، لومیکان نقش کنترل قطر فیبرهای کلاژن را در حدود ۲۵ نانومتر بر عهده دارد، در حالی که کراتوکان و میمکان فاصله بین فیبری را یکنواخت نگه میدارند. هنگامی که زنجیرههای جانبی سولفاته این SLRPها کوتاه و ناقص هستند، فیبرهای کلاژن در ضخامت دچار ناهمگنی شده و فاصله بین آنها نیز نامنظم میشود. در نتیجه، پراکندگی نور در استرومای قرنیه افزایش یافته و از نظر بالینی به صورت کدورت شیشهای (مات) مشاهده میشود.
برای حفظ شفافیت قرنیه، نظریه شبکه موریس (Maurer) به طور کلاسیک شناخته شده است که بر اساس آن «آرایش منظم شبکهای فیبرهای کلاژن باعث خنثیسازی تداخل نوری میشود». در دیستروفی قرنیه ماکولار (MCD)، این ساختار شبکهای به دلیل ناهنجاری SLRP مختل شده و شفافیت از بین میرود2,4).
مطالعات پایه اخیر نشان دادهاند که اختلال در عملکرد اتوفاژی ناشی از جهش CHST6 ممکن است باعث پیروپتوزیس (مرگ سلولی التهابی) کراتوسیتها شده و به پیشرفت بیماری کمک کند8). ناهنجاری مشابه اتوفاژی در سایر دیستروفیهای قرنیه (مانند نوع II دانهای) نیز گزارش شده است و به عنوان یک پاتوژنز مشترک در دیستروفیهای قرنیه مورد توجه قرار گرفته است.
ژندرمانی هدفمند به عنوان یک استراتژی درمانی دائمی پیشنهاد شده است 8). تحقیقات پایهای ویرایش ژن با استفاده از CRISPR/Cas9 در دیستروفی اپیتلیال قرنیه میزمن پیشرو بوده و ممکن است در آینده گزینه درمانی برای MCD باشد. با این حال، چالشهای زیادی برای کاربرد بالینی باقی مانده است، از جمله ویرایش ناخواسته آلل طبیعی (اثرات خارج از هدف)، ایجاد روش کارآمد انتقال ژن به سلولهای استرومای قرنیه و تأیید ایمنی طولانیمدت.
رویکرد حذف آنزیمی کراتان سولفات کم سولفاته انباشته شده در قرنیه به صورت پایهای بررسی شده است. این روش مفهوم درمان جایگزینی آنزیم را که در موکوپلی ساکاریدوز سیستمیک پیشرو است، به صورت موضعی در قرنیه اعمال میکند، اما تاکنون گزارشی از کاربرد بالینی آن منتشر نشده است.
پیوند اندوتلیال قرنیه با برداشتن غشای دسمه (DSAEK) و پیوند اندوتلیال قرنیه با غشای دسمه (DMEK) عمدتاً برای دیستروفی اندوتلیال قرنیه فوکس و کراتوپاتی تاولی توسعه یافتهاند، اما کاربرد آنها در موارد MCD با درگیری اولیه اندوتلیوم نیز موضوع تحقیقات آینده است. در حال حاضر، برای این بیماری که هم استروما و هم اندوتلیوم را درگیر میکند، پیوند نافذ قرنیه (PKP) گزینه عملی است. در آینده، مفهوم «پیوند متوالی» با ترکیب مرحلهای پیوند لایهای عمیق قدامی (DALK) برای جایگزینی استروما و DMEK برای جایگزینی اندوتلیوم پیشنهاد شده است، اما همه اینها در مرحله تحقیقاتی هستند.
غلظت کراتان سولفات سرم در بیماران MCD با ایمونوفنوتیپ نوع II کمتر از نرمال گزارش شده است و کاربرد آن به عنوان یک نشانگر متابولیک سیستمیک مورد بحث است 2). در آینده، انتظار میرود که از آن برای غربالگری با آزمایش خون و تشخیص زودهنگام ناقلین در خانوادههای دارای جهش CHST6 استفاده شود. علاوه بر این، تجزیه و تحلیل خودکار تصاویر لامپ شکاف با هوش مصنوعی نیز حوزه تحقیقاتی است که میتواند به تشخیص زودهنگام بیماریهای نادر مانند این کمک کند.
از آنجایی که این بیماری با وراثت اتوزومال مغلوب منتقل میشود، حدود ۲۵٪ از خواهران و برادران بیمار ممکن است مبتلا شوند. با استفاده از آزمایش ژن CHST6، غربالگری خانوادگی و مشاوره ژنتیک در مورد ازدواج و بارداری امکانپذیر است. در خانوادههایی با سابقه ازدواج فامیلی یا موارد مشابه کدورت قرنیه، ارجاع زودهنگام به متخصص توصیه میشود.
Akama TO, Nishida K, Nakayama J, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nature Genetics. 2000;26(2):237-241. doi:10.1038/79987.
Aggarwal S, Peck T, Golen J, Karcioglu ZA. Macular corneal dystrophy: A review. Survey of Ophthalmology. 2018;63(5):609-617. doi:10.1016/j.survophthal.2018.03.004.
Jonasson F, Oshima E, Thonar EJ, et al. Macular corneal dystrophy in Iceland. A clinical, genealogic, and immunohistochemical study of 28 patients. Ophthalmology. 1996;103(7):1111-1117. doi:10.1016/s0161-6420(96)30559-9.
Jonasson F, Johannsson JH, Garner A, Rice NS. Macular corneal dystrophy in Iceland. Eye (London, England). 1989;3 ( Pt 4):446-54. doi:10.1038/eye.1989.66. PMID:2606219.
Sultana A, Sridhar MS, Jagannathan A, et al. Novel mutations of the carbohydrate sulfotransferase-6 (CHST6) gene causing macular corneal dystrophy in India. Mol Vis. 2003;9:730-734. PMID:14735064.
Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating keratoplasty for macular corneal dystrophy. Ophthalmology. 2005;112(2):220-224. doi:10.1016/j.ophtha.2004.08.017.
Reinhart WJ, Musch DC, Jacobs DS, Lee WB, Kaufman SC, Shtein RM. Deep anterior lamellar keratoplasty as an alternative to penetrating keratoplasty a report by the american academy of ophthalmology. Ophthalmology. 2011;118(1):209-218. doi:10.1016/j.ophtha.2010.11.002. PMID:21199711.
متن مقاله را کپی کنید و در دستیار هوش مصنوعی دلخواه خود بچسبانید.
مقاله در کلیپبورد کپی شد
یکی از دستیارهای هوش مصنوعی زیر را باز کنید و متن کپیشده را در کادر گفتگو بچسبانید.