Macular corneal dystrophy (MCD) is a hereditary corneal dystrophy in which glycosaminoglycans (mainly keratan sulfate) accumulate in the corneal stroma. It follows an autosomal recessive inheritance pattern and is caused by mutations in the CHST6 gene located on the long arm of chromosome 16 (16q22)1,3). It was formerly called Groenouw corneal dystrophy type II or Fehr corneal dystrophy.
While many other corneal stromal dystrophies (granular, lattice) are autosomal dominant, this disease is characterized by autosomal recessive inheritance. In Japan, it is considered one of the four major corneal dystrophies along with granular corneal dystrophy (types I and II), lattice corneal dystrophy (types I and IIIA), and gelatinous drop-like corneal dystrophy, accounting for about 96% of all corneal dystrophies. Among these, the first two are autosomal dominant, while the latter two (gelatinous drop-like and macular) are autosomal recessive.
In the IC3D (International Committee for Classification of Corneal Dystrophies) classification, MCD is positioned as a type of stromal dystrophy 1). In a 2018 review by Aggarwal et al. in Survey of Ophthalmology, this disease is detailed as “a rare but visually significant stromal dystrophy,” and the diagnostic and treatment system is summarized 4). In the second edition of the IC3D classification, corneal dystrophies are classified into categories 1 to 4, distinguished by the strength of evidence for causative genes, pathological findings, and clinical features 1). MCD is classified as Category 1 (a dystrophy established at the genetic level) due to the identification of CHST6 gene mutations.
Historically, this disease was first described by Groenouw in 1890, and later a convention emerged calling granular dystrophy “type I” and macular dystrophy “type II.” In 1938, Jones and Zimmerman established it as an independent disease, and in 2000, Akama et al. identified the CHST6 gene, clarifying its molecular basis 3).
Worldwide, there are large regional differences, and in areas with high prevalence, familial clustering is observed. It is a relatively rare disease.
Regional Differences in Prevalence
United States: Rare, approximately 0.3 per 250,000 people 2,3)
Iceland: Approximately 19 per 250,000 people, one of the highest frequencies in the world 5,6)
High prevalence regions: South India, Saudi Arabia, Iceland, and Northern Europe have high frequency 5,7)
Other regions: Relatively rare. Occurs with consanguineous marriage or compound heterozygosity.
Immunophenotypes
Type I: Keratan sulfate negative in both cornea and serum 2)
Type IA: Positive in corneal keratocytes, negative in serum 2)
Type II: Keratan sulfate positive in both cornea and serum 2)
Clinical features: All three types have identical phenotypes and cannot be distinguished by slit lamp2,8)
The immunophenotypes of MCD are classified by the amount of keratan sulfate in the cornea and serum using anti-keratan sulfate monoclonal antibodies 2,3).
Phenotype
Corneal keratan sulfate
Serum keratan sulfate
Type I
Negative
Negative
Type IA
Positive (intracellular)
Negative
Type II
Positive
Positive
Most patients are classified as type I or IA. However, clinically, distinguishing these subtypes is not important, and they cannot be differentiated by examination findings 2,8).
QHow is macular corneal dystrophy different from other corneal dystrophies?
A
The biggest difference is that it follows an autosomal recessive inheritance pattern. Granular and lattice corneal dystrophies are autosomal dominant, but this disease requires mutations in both alleles of the CHST6 gene. It also presents with diffuse ground-glass opacities, causing the entire cornea to become cloudy, and slit-lamp examination reveals deposits in the superficial layer centrally and in the deep layer peripherally.
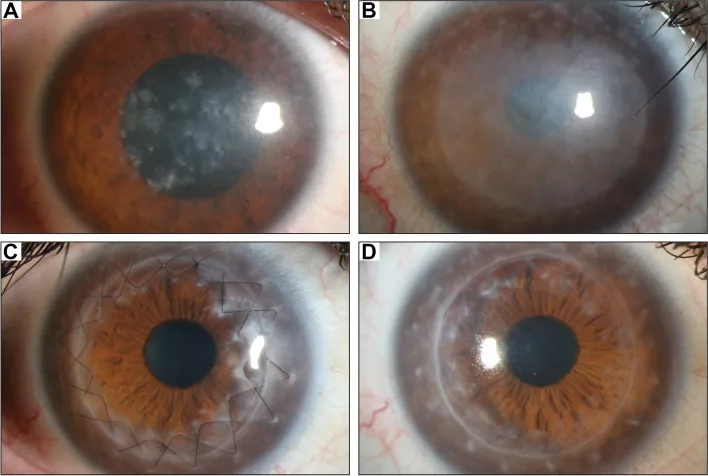
Gassel CJ, et al. Histological findings of corneal tissue after failed phototherapeutic keratectomy in macular corneal dystrophy - a case report. BMC Ophthalmol. 2022. Figure 1. PMCID: PMC9080147. License: CC BY.
Slit-lamp photograph shows diffuse gray-white opacities and macular deposits from the central cornea to the entire cornea. This represents typical clinical findings of macular corneal dystrophy, making it easy to understand the corneal opacities that cause vision loss.
The typical progression pattern of clinical findings is as follows.
Clinically, fine deposits are seen diffusely in the corneal stroma, causing a ground-glass opacity. As it progresses, the opacity involves the full thickness of the stroma and spreads from the center to the periphery. Subsequently, in addition to the faint opacity, numerous small, irregular gray-white opacities appear in the superficial to deep stroma. 9)
Early Findings
Spotty opacities: Small gray-white spotty opacities appear in the superficial stroma of the central cornea
Ground-glass opacity: Diffuse faint opacity is seen in the corneal stroma
Indistinct borders: The margins of the opacities are unclear, and the boundary with normal stroma is indistinct
Advanced Stage Findings
Full-thickness involvement: Opacification extends through the entire stroma
Peripheral extension: Opacification spreads from the center to the periphery
Corneal thinning: Central corneal thickness decreases
Endothelial and Descemet’s membrane deposits: Abnormal material accumulates in deep structures
On slit-lamp examination, the entire cornea appears diffusely opaque with irregular gray-white patchy deposits. Optical sectioning with a slit beam reveals a characteristic distribution: deposits are located in the superficial layer centrally and in the deep layer peripherally. The patchy lesions often appear in a concentric pattern8).
Opacification may extend to the limbus, which is an important distinguishing feature from other corneal dystrophies. In granular corneal dystrophy and lattice corneal dystrophy, the limbus often remains clear, whereas in MCD, the entire cornea including the limbus is frequently opaque2,8). Irregular astigmatism is associated with anterior stromal deposits, and decreased corneal sensation may also be present. Since abnormal material deposits on the endothelium, advanced cases may develop stromal edema due to endothelial dysfunction8).
The natural course varies among individuals but often follows these stages:
Early childhood (asymptomatic stage): The genetic mutation is present from birth, but slit-lamp findings are minimal and the patient is asymptomatic.
School age to adolescence (early opacification stage): Diffuse faint opacities appear in the superficial corneal stroma, and eventually patchy deposits become visible.
Ages 10–30 (vision decline stage): Opacification progresses, and patients become aware of decreased vision.
Ages 30–40 (advanced stage): Opacification spreads to the full thickness of the stroma and to the limbus, with clear corneal hypoesthesia, thinning, and irregular astigmatism.
Middle to older age (corneal transplant indication stage): Visual impairment reaches a level that interferes with daily life, and transplantation is considered.
MCD is a progressive disease with continuous decline in visual function throughout life, which differs significantly in prognosis from granular corneal dystrophy type I and some subtypes of lattice corneal dystrophy, where visual impairment remains mild4,8).
The causative gene is CHST6 (carbohydrate sulfotransferase 6)3). It is located on 16q22 and encodes an enzyme that transfers sulfate groups to N-acetylglucosamine on corneal proteoglycans. Many types of mutations in this gene have been reported in various ethnic groups, including missense mutations, nonsense mutations, frameshift mutations, and deletions in the 5’ upstream region3,7).
Because it is an autosomal recessive disorder, both parents of an affected individual are usually carriers. The incidence tends to be higher in regions or populations with frequent consanguineous marriages. Compound heterozygotes resulting from marriages between different families can also develop the disease7).
In 2000, Akama et al. identified CHST6 as the causative gene for this disease and showed that both immunophenotypes I and II are caused by mutations in the same gene locus3). This discovery was an important finding suggesting that differences in immunophenotype are determined by different mutation patterns in a single gene, and it became the basis for subsequent genetic diagnostic systems.
More than 200 types of mutations have been reported, with missense mutations being the most common. Sultana et al. identified many novel mutations in a group of patients from South India, showing that the high frequency in that region is due to an accumulation of region-specific mutations7). Similar regional clustering has been reported in Saudi Arabia and Iceland, and it is thought that the historical background of the population (founder effects and consanguineous marriage customs) contributes to the incidence5,6,7).
The incidence varies by region, and the disease is relatively rare9). Compared to other corneal dystrophies such as granular corneal dystrophy, there are fewer cases, and it tends to be reported in families with compound heterozygosity or a background of consanguineous marriage.
Family history: Because it is autosomal recessive, both parents must be carriers.
Consanguineous marriage: Increases the incidence.
Geographic region: Higher prevalence in South India, Saudi Arabia, Iceland, and Scandinavia5,7).
QIs genetic testing necessary?
A
Genetic testing of the CHST6 gene is useful for definitive diagnosis. Genetic testing can be performed at accredited medical institutions. However, in many facilities, clinical diagnosis using slit-lamp microscopy is the main method. It is useful for assessing the risk of disease in future children and for confirming diagnosis in cases with atypical clinical findings.
The trigger to suspect MCD is “bilateral, progressive, diffuse corneal opacity in adolescents to young adults.” First, a detailed history of subjective symptoms (decreased vision, photophobia, irritation), family history, and consanguineous marriage is obtained. Next, the standard approach includes slit-lamp microscopic evaluation of corneal findings, endothelial function assessment, and genetic testing if necessary.
This is the basic examination for diagnosis. If bilateral corneal opacity without congestion or corneal edema is observed, corneal dystrophy should be suspected. In MCD, the following findings are characteristic:
Diffuse ground-glass opacity: The entire cornea appears diffusely cloudy
Concentric distribution: On slit beam, central opacities are superficial, peripheral ones are deep
Limbus involvement: Opacities may extend to the limbus
Endothelial abnormalities: In advanced cases, guttata-like deposits may be seen
While many corneal dystrophies appear as discontinuous lesions (with clear areas between deposits) at the slit-lamp level, MCD exceptionally presents a diffuse opacity pattern. Along with lattice corneal dystrophy type I and gelatinous drop-like corneal dystrophy, it is characterized as a representative example of “diffusely observed corneal deposits.”
Specular microscope: Evaluation of endothelial cell density and morphology. The extent of endothelial pathology directly influences surgical technique selection
Corneal Pentacam (Scheimpflug imaging): Provides a density map of the entire cornea, useful for three-dimensional evaluation of opacity sites
Histologically, it shows positivity with Alcian blue and colloidal iron staining, and diffuse accumulation of undersulfated glycosaminoglycans within and around keratocytes in the corneal stroma is observed2,8). Breaks in Bowman’s layer may be seen, and in advanced cases, abnormal material is also found within endothelial cells. Guttata-like findings on Descemet’s membrane may also be present.
Other conditions to consider include posterior amorphous corneal dystrophy (PACD) and pre-Descemet corneal dystrophy (PDCD). Systemic mucopolysaccharidoses (such as Hurler, Scheie, and Morquio syndromes) can also present with corneal opacification, so evaluation including systemic findings is necessary 8).
QHow is macular corneal dystrophy diagnosed?
A
Clinical diagnosis is made by slit-lamp microscopy showing diffuse ground-glass corneal opacity and macular deposits, bilateral and progressive nature, family history, and age of onset (10–30 years). CHST6 genetic testing is useful for definitive diagnosis.
The goals of treatment for MCD are: (1) maintenance and recovery of visual function, (2) alleviation of pain and irritation symptoms through stabilization of the ocular surface, and (3) prevention of complications (epithelial erosion, infection). Since there is no fundamental etiological treatment, staged interventions are performed according to the disease stage and symptoms.
For cases with progressive visual loss, corneal transplantation is the only curative option. The surgical technique is chosen based on the presence or absence of endothelial involvement.
Deep anterior lamellar keratoplasty (DALK): This is the first choice for cases without endothelial involvement 8,9). By preserving the patient’s own corneal endothelium, the risk of graft rejection is lower than with penetrating keratoplasty. The American Academy of Ophthalmology (AAO) assessment report also states that for stromal dystrophies, DALK achieves visual recovery equivalent to PKP with less endothelial loss 8). The recurrence rate after DALK is reported to be low, and recurrence is unlikely because the recipient’s corneal stroma is replaced.
Penetrating keratoplasty (PKP): This is indicated for advanced cases with abnormal deposits involving the endothelium and Descemet’s membrane, and for cases with severe central corneal thinning 7). The average age for first PKP for MCD is reported to be in the 30s to 40s 7), and graft survival is good.
Phototherapeutic keratectomy (PTK): This is performed symptomatically for recurrent corneal epithelial erosion and superficial scarring opacities. However, caution is needed regarding induced hyperopia and stromal haze.
In MCD, abnormal deposits can also involve endothelial cells, so PKP tends to be chosen over DALK when the disease extends deep7). Full-thickness corneal transplantation is indicated when endothelial abnormalities are present. Detailed preoperative endothelial evaluation (specular and confocal microscopy) is key to determining the surgical technique.
The recurrence rate after DALK is reported to be low, approximately 5%. Graft survival after PKP is good, with many reports achieving long-term survival; however, there are also reports of abnormal material redepositing on the graft several to more than ten years after surgery 8). In a series from Saudi Arabia by Al-Swailem et al., graft survival after PKP for MCD was good, but recurrence was observed in some cases during long-term follow-up 8). Additionally, the AAO review by Reinhart et al. showed that DALK achieves visual recovery equal to or better than PKP and low endothelial cell loss for stromal dystrophies 9). A multicenter study by Unal et al. also reported the efficacy of DALK for stromal dystrophies including MCD.
In general, postoperative recurrence of corneal grafts occurs because the molecular pathology of the original disease remains in the host. In DALK, the host endothelium and pre-Descemet layer are preserved, so in cases with endothelial involvement, the disease may progress. Long-term regular follow-up (visual acuity, slit-lamp examination, endothelial cell density measurement) is necessary after surgery.
QWhich should I choose, DALK or PKP?
A
If the endothelium and Descemet membrane are not involved, deep anterior lamellar keratoplasty (DALK) is the first choice. DALK preserves the patient’s own corneal endothelium, so the risk of rejection is low, and the postoperative recurrence rate is reported to be about 5%. On the other hand, in cases where abnormal deposits also involve the endothelium, penetrating keratoplasty (PKP) is indicated. The surgical procedure is determined based on preoperative endothelial evaluation.
The CHST6 gene encodes carbohydrate sulfotransferase 6 3). This enzyme is responsible for transferring sulfate groups to N-acetylglucosamine on keratan molecules and is essential for the normal synthesis of keratan sulfate (KS) in corneal proteoglycans.
When enzyme activity is lost due to gene mutation, undersulfated keratan sulfate is synthesized. This abnormal keratan sulfate has low solubility and accumulates abnormally inside and outside keratocytes in the corneal stroma2,3).
Quantitative and qualitative abnormalities of keratan sulfate lead to the following chain of pathological changes.
Abnormal production of small leucine-rich proteoglycans (SLRPs): Corneal-specific SLRPs such as lumican, keratocan, and mimecan are not synthesized normally.
Abnormal arrangement of collagen fibers: These SLRPs tightly control the diameter and spacing of corneal collagen fibers, ensuring transparency. Reduced SLRP function leads to uneven collagen fiber diameters and altered fiber spacing2).
Abnormal accumulation in the extracellular matrix: Unsulfated keratan itself deposits in the extracellular matrix.
Increased light scattering and loss of transparency: The combined changes above increase scattering of visible light, causing diffuse opacity throughout the cornea.
Accumulation of glycosaminoglycans is observed both inside and outside keratocytes in the stroma, and as the disease progresses, it spreads to Bowman’s layer, Descemet’s membrane, and endothelial cells. In type I, reduced enzyme activity has also been confirmed in auricular cartilage, suggesting it may be a partial manifestation of systemic keratan sulfate metabolism abnormality2). However, systemic symptoms are rarely seen clinically, and it is treated as a localized disease primarily affecting the cornea.
Among corneal proteoglycans, lumican controls collagen fiber diameter to about 25 nm, while keratocan and mimecan maintain uniform fiber spacing. When the sulfated side chains of these SLRPs are short and incomplete, collagen fibers become variable in thickness and spacing becomes uneven. As a result, light scattering within the corneal stroma increases, clinically observed as ground-glass opacity.
Classically, the Maurer lattice theory explains corneal transparency by the cancellation of light interference due to the orderly lattice arrangement of collagen fibers. In MCD, this lattice structure is disrupted by SLRP abnormalities, leading to loss of transparency2,4).
Recent basic research has reported that impaired autophagy due to CHST6 mutations may induce pyroptosis (inflammatory cell death) of keratocytes, contributing to disease progression8). Similar autophagy abnormalities have been reported in other corneal dystrophies (e.g., granular type II), drawing attention as a common pathology in corneal dystrophies.
Gene-targeted therapy has been proposed as a permanent treatment strategy 8). Basic research on gene editing using CRISPR/Cas9 is progressing in Meesmann corneal epithelial dystrophy, and it may become a future treatment option for MCD as well. However, many challenges remain for clinical application, including unintended editing of normal alleles (off-target effects), establishment of efficient gene delivery methods to keratocytes, and verification of long-term safety.
An approach to enzymatically remove accumulated undersulfated keratan sulfate in the cornea is being investigated at a basic level. This applies the concept of enzyme replacement therapy, which is already used in systemic mucopolysaccharidoses, to the local cornea, but there are currently no reports of clinical application.
Descemet stripping automated endothelial keratoplasty (DSAEK) and Descemet membrane endothelial keratoplasty (DMEK) are surgical techniques developed mainly for Fuchs endothelial corneal dystrophy and bullous keratopathy, but their application to cases of MCD with early endothelial involvement is a topic for future research. Currently, PKP is the realistic choice for this disease, which affects both stroma and endothelium. In the future, the concept of “sequential keratoplasty” combining DALK (replacing only the stroma) and DMEK (replacing only the endothelium) in stages has been proposed, but these are still at the research stage.
Serum keratan sulfate levels are reported to be lower than normal in patients with immunophenotype II MCD, and their usefulness as a systemic metabolic marker is being discussed 2). In the future, application to blood test screening and early carrier diagnosis in families with CHST6 mutations is expected. Furthermore, automated analysis of slit-lamp images using artificial intelligence is also a research area that may contribute to early detection of rare diseases such as this one.
Since this is an autosomal recessive disorder, approximately 25% of the patient’s siblings may be affected. CHST6 genetic testing enables family screening and genetic counseling regarding marriage and pregnancy. Early referral to a specialist is recommended for families with a history of consanguineous marriage or cases of similar corneal opacity within the family.
Akama TO, Nishida K, Nakayama J, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nature Genetics. 2000;26(2):237-241. doi:10.1038/79987.
Aggarwal S, Peck T, Golen J, Karcioglu ZA. Macular corneal dystrophy: A review. Survey of Ophthalmology. 2018;63(5):609-617. doi:10.1016/j.survophthal.2018.03.004.
Jonasson F, Oshima E, Thonar EJ, et al. Macular corneal dystrophy in Iceland. A clinical, genealogic, and immunohistochemical study of 28 patients. Ophthalmology. 1996;103(7):1111-1117. doi:10.1016/s0161-6420(96)30559-9.
Jonasson F, Johannsson JH, Garner A, Rice NS. Macular corneal dystrophy in Iceland. Eye (London, England). 1989;3 ( Pt 4):446-54. doi:10.1038/eye.1989.66. PMID:2606219.
Sultana A, Sridhar MS, Jagannathan A, et al. Novel mutations of the carbohydrate sulfotransferase-6 (CHST6) gene causing macular corneal dystrophy in India. Mol Vis. 2003;9:730-734. PMID:14735064.
Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating keratoplasty for macular corneal dystrophy. Ophthalmology. 2005;112(2):220-224. doi:10.1016/j.ophtha.2004.08.017.
Reinhart WJ, Musch DC, Jacobs DS, Lee WB, Kaufman SC, Shtein RM. Deep anterior lamellar keratoplasty as an alternative to penetrating keratoplasty a report by the american academy of ophthalmology. Ophthalmology. 2011;118(1):209-218. doi:10.1016/j.ophtha.2010.11.002. PMID:21199711.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.