Granular corneal dystrophy (GCD) is a hereditary corneal disease characterized by granular deposits in the corneal stroma. In the second edition of the International Classification of Corneal Dystrophies (IC3D 2015), it is classified as an epithelial-stromal TGFBI-related dystrophy 2).
It is caused by point mutations in the TGFBI gene (chromosome 5q31) and follows an autosomal dominant inheritance pattern. Based on the mutation, it is classified into the following two types.
Classification
Main Mutation
Alternative Name / Former Name
Main Deposits
GCD1
Arg555Trp (R555W)
Classic granular / Groenouw type 1
Hyaline only
GCD2
Arg124His (R124H)
Avellino corneal dystrophy
Hyaline + amyloid
GCD2 was reported in 1988 as a subtype independent from GCD1, and was called Avellino corneal dystrophy because the first family originated from the Avellino region of Italy 1). Later, in 1997, the causative gene TGFBI was identified and the R124H mutation was found 3). Since the second edition of the IC3D classification (2015), it has been officially named granular corneal dystrophy type 2 (GCD2), with “Avellino” listed as a historical name 2).
Granular corneal dystrophy is internationally classified under the epithelial-stromal dystrophies group. This group includes six TGFBI-related diseases: granular (GCD1, GCD2), lattice (LCD1, LCD3A), Reis-Bücklers, and Thiel-Behnke, all caused by different point mutations in the TGFBI gene on 5q31 2,4). In Japanese ophthalmic practice, these six diseases are collectively referred to as “TGFBI-related corneal dystrophies.”
Granular corneal dystrophy was first reported by Groenouw in 1890 and was initially called “Groenouw type 1.” In 1938, it was clearly distinguished from lattice dystrophy, and for a long time it was treated as a single disease entity. In 1988, a family from the Avellino region of Italy was reported with features of both granular and lattice dystrophies, which was later separated as GCD2 (Avellino type) 1,2). The current classification system was established with the 2015 revision of the IC3D classification, and genotype-based naming became the international standard 2).
Inheritance pattern: Autosomal dominant with high penetrance
Type 1: Common in Europe and the United States; rare in Japan
Type 2: Overwhelmingly common in East Asia, such as Japan and South Korea. The prevalence in South Korea is about 11.5 per 10,000 people 1)
Proportion among TGFBI-related corneal dystrophies: GCD2 accounts for 72–91% in South Korea and Japan, 36% in the United States, and 3% in Poland 1)
Japanese genetic diagnosis data: At Yamaguchi University, 234 corneal dystrophy patients underwent genetic diagnosis over 21 years (2000–2021), and the four major corneal dystrophies (granular type I and II, lattice type I and IIIA, gelatinous drop-like, and macular) accounted for about 96% of all cases. - Characteristics in East Asia: Granular corneal dystrophy is overwhelmingly type 2 (R124H) in East Asia. - Age of onset: Heterozygous GCD2 shows micro-opacities observable only by slit lamp from school age, but no subjective symptoms. Subjective visual decline typically appears in the 40s–50s.
Sex difference: Autosomal dominant inheritance, no sex difference.
QWhat does "granular" refer to?
A
It refers to a condition where many small, well-demarcated white to gray-white clumps (granular deposits) form in the superficial stroma of the central cornea. When observed directly with a slit-lamp microscope, they are described as breadcrumb-like, snowflake-like, or candy-like. The deposits originate from mutant TGFBI protein.
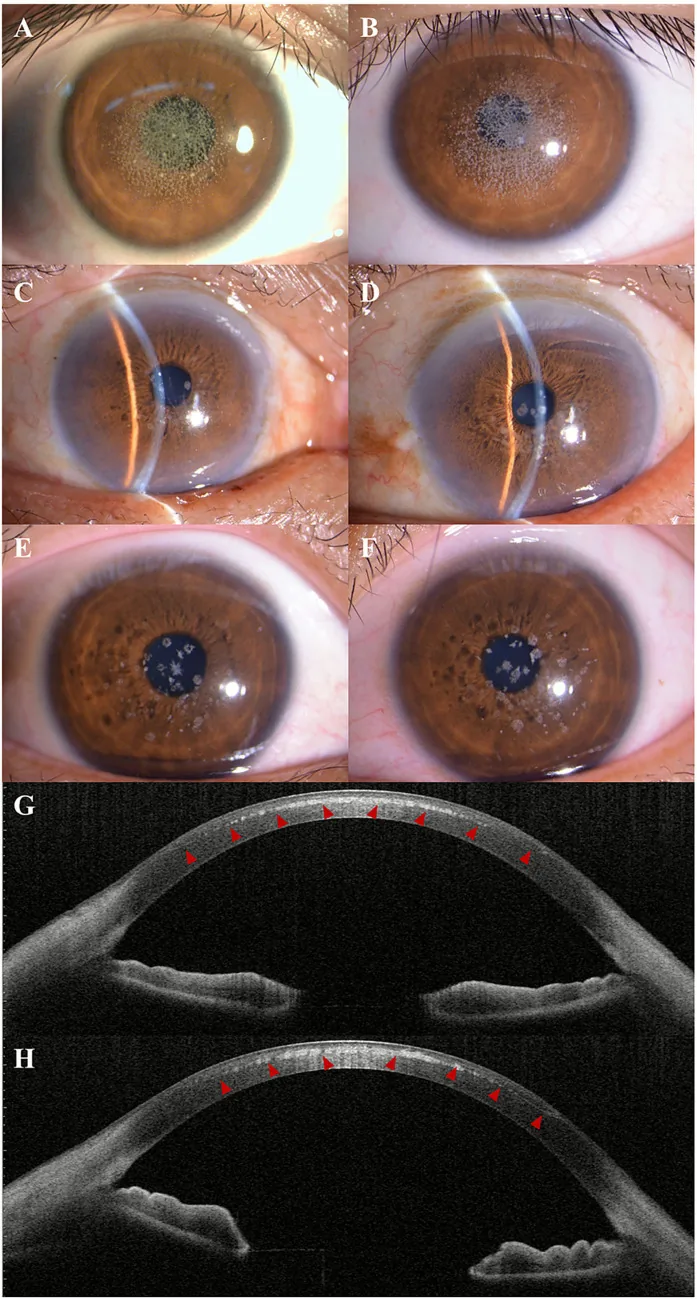
Kuang L, et al. Case Report: Post-LASIK exacerbation of granular corneal dystrophy type 2: a familial case with TGFBI mutation. Front Med (Lausanne). 2025. Figure 1. PMCID: PMC12722859. License: CC BY.
Slit-lamp photograph shows scattered and clustered gray-white granular opacities in the central to paracentral cornea. AS-OCT shows hyperreflective deposits in the anterior corneal stroma, indicating clinical findings of granular corneal dystrophy.
Asymptomatic to mild: In heterozygotes, early to intermediate stages often have no subjective visual decline, and many cases are discovered incidentally during checkups. Most patients complain of visual decline in their 40s–50s.
Glare and photophobia: When opacities involve the pupillary area, patients complain of daytime glare and reduced contrast sensitivity.
Recurrent corneal epithelial erosion: Deposits damage Bowman’s layer and the epithelial basement membrane, causing sharp eye pain, tearing, and redness during sleep or upon waking.
Visual decline: When the transparent areas between deposits become opaque, vision progressively decreases3).
Reduced contrast sensitivity: Contrast sensitivity often decreases before visual acuity (Landolt ring test) declines.
Day blindness tendency: Because scattered light strongly affects vision in bright conditions, patients complain of glare outdoors or while driving.
Difficulty correcting with glasses or contact lenses: Scattering from deposits cannot be improved by refractive correction.
In homozygotes, marked visual impairment occurs from early childhood (4–7 years of age), and treatment is required around age 10.
Clinical Findings (Findings Confirmed by Physician Examination)
Granular opacities: Relatively small, well-demarcated white to gray-white granular opacities scattered in the central cornea. Described as breadcrumb-like or snowflake-like.
Depth: Subepithelial and superficial corneal stroma. Does not extend to the limbus.
Deposits: Hyaline only. Stains red with Masson’s trichrome. Does not contain amyloid.
Progression: The number of granules increases with age, and the borders become indistinct.
GCD2 (R124H)
Granular opacities: Onset with larger white to gray-white well-demarcated opacities than in GCD1. Phenotypes are diverse, including confetti-like, linear, stellate, and rod-shaped.
Mixed type: Fine lattice-like linear opacities seen in lattice corneal dystrophy may also be present.
Deposits: Both hyaline and amyloid. Masson’s trichrome positive and Congo red positive, exhibiting yellow-green birefringence under polarized light.
Progression: After age 25–30, dense white rod-shaped and stellate opacities appear in the mid-stroma. Diffuse sheet-like deposits become prominent in the superficial layer, making PTK a good indication3).
In both types, opacities are located in the central cornea and do not extend to the limbal periphery. Usually bilateral with little asymmetry.
Homozygous mutations result in markedly different phenotypes.
GCD1 homozygotes: White reticular opacities at the same depth from the subepithelium to the superficial stroma, with almost no gaps. As it progresses, the iris and anterior chamber become unobservable.
GCD2 homozygotes: Solid round white opacities appear over the entire cornea except the periphery, with no gaps. It is severe enough to be recognized by the naked eye, and only the limbal transparency is preserved3). Homozygous cases are refractory corneal dystrophies that recur within a short period of 1–2 years even after PTK or corneal transplantation.
QHow does the course differ between homozygotes and heterozygotes?
A
Homozygotes develop in early childhood (4–7 years) and progress rapidly. White opacities appear over the entire cornea with no gaps, and PTK or corneal transplantation is required around age 10. Even after surgery, it recurs within 1–2 years, following a refractory course. Heterozygotes progress slowly and can usually maintain good vision until their 40s–50s.
GCD is caused by point mutations in the TGFBI gene (chromosome 5q31). The TGFBI gene encodes TGFBIp (keratoepithelin), an extracellular matrix protein. Mutant TGFBIp has reduced susceptibility to proteolysis and accumulates as abnormal insoluble deposits in the corneal stroma1,5,7).
The TGFBI-related corneal dystrophies include the following2,4):
Granular corneal dystrophy type 1 (R555W)
Granular corneal dystrophy type 2 (R124H, formerly Avellino)
GCD is an autosomal dominant disorder with high penetrance. If a proband is diagnosed, 50% of first-degree relatives (parents, siblings, children) may carry the same mutation. Early identification of asymptomatic carriers in the family can help avoid future refractive surgery and establish regular follow-up for progression monitoring 1,5). Especially in young individuals considering LASIK, careful family history taking and genetic testing when indicated are strongly recommended.
Clinical diagnosis is based on slit-lamp examination showing well-demarcated granular opacities in the anterior stroma and a positive family history. Corneal dystrophy should be suspected when bilateral corneal opacities (deposits) are present without injection or corneal edema3).
In the differential diagnosis of corneal dystrophies, first determine whether the deposits are “well-demarcated” or “diffuse” 3). For well-demarcated granular deposits, differentiate GCD1 (small) from GCD2 (large) based on deposit size. In GCD2, scleral scatter reveals diffuse sheet-like opacities between the granular deposits; these sheet-like opacities are a good indication for PTK3).
Slit-lamp microscopy: Direct observation of well-demarcated white granular opacities. Also use scleral scatter, retroillumination, and transillumination.
Corneal topography: Provides additional information on the density of opacities.
Genetic testing: TGFBI gene analysis is useful for definitive diagnosis. In Japan, it has been covered by insurance as a corneal dystrophy genetic test since April 2020 3).
Anterior segment photography: For long-term follow-up, it is important to take high-quality anterior segment photographs at the initial visit and at each follow-up, and to keep them in the medical record.
GCD1: Hyaline deposits that stain red with Masson’s trichrome stain. Does not contain amyloid. Electron microscopy shows rod-shaped or trapezoidal deposits.
GCD2: Both hyaline (Masson’s trichrome positive) and amyloid (Congo red positive, apple-green birefringence under polarized light) are deposited. Electron microscopy reveals rod-shaped electron-dense deposits and amyloid fibrils 1,7).
Lattice corneal dystrophy type 1 (LCD1): TGFBI R124C mutation. Linear and lattice-shaped opacities due to amyloid deposition in the stroma. Often associated with recurrent corneal epithelial erosion3)
Macular corneal dystrophy (MCD): CHST6 gene mutation. Autosomal recessive. Diffuse opacification of the entire cornea
Fleck corneal dystrophy (FCD): PIP5K3 mutation. Small white spots throughout the stroma, usually asymptomatic
QIs genetic testing covered by insurance?
A
Since April 2020, genetic testing for corneal dystrophy has been covered by insurance. However, facility certification is required, so the number of facilities that can perform the test is limited. If clinical findings are suspicious or if refractive surgery such as LASIK is being considered, definitive diagnosis by genetic testing is desirable.
In the early stage without visual impairment or recurrent corneal epithelial detachment, treatment is not necessary. Surgical intervention is considered when visual impairment extends to the pupillary area.
Artificial tears: Use sodium hyaluronate 0.1% or 0.3% eye drops 4 to 6 times a day to reduce dryness and irritation
Therapeutic soft contact lenses: For recurrent epithelial detachment, protect the ocular surface and promote healing. Generally worn all day, requiring regular replacement
Antibiotic eye drops/ointment: To prevent secondary infection during epithelial detachment, use levofloxacin 0.5% eye drops 3 to 4 times a day and ofloxacin eye ointment at bedtime
Hypertonic saline (5% sodium chloride eye drops/ointment): May be used as an adjunctive treatment to reduce epithelial edema.
GCD2 homozygotes: Recurrence occurs approximately 18 months after the first PTK, and after the second or third procedure, recurrence occurs within about 3 months1).
GCD2 heterozygotes: Recurrence after PTK is relatively slow, with an average of 38.4 months1).
Combined use of mitomycin C (MMC): MMC use during PTK is not recommended. MMC induces apoptosis of keratocytes in the corneal stroma, reducing cells responsible for reabsorption and degradation of TGFBIp, which may accelerate recurrence1).
Cases of exacerbation after LASIK: PTK can be performed, but the effect is greater after removal of the LASIK flap1,8).
Postoperative management: After PTK, use antibacterial eye drops (levofloxacin 0.5%) and corticosteroid eye drops (fluorometholone 0.1%) four times daily until epithelial healing, then taper. Epithelial healing usually takes 3 to 5 days.
DALK (deep anterior lamellar keratoplasty) in practice
DALK is a procedure that preserves the endothelium. Using the big bubble technique, the stroma is dissected and removed up to just above Descemet’s membrane, and donor stroma is sutured. Since there is no risk of endothelial rejection, long-term prognosis is considered better than PK3). In a study by Kitazawa et al., visual acuity at 5 years after DALK for TGFBI-related corneal dystrophies (including granular and lattice types) was generally good, and graft survival rate was high10). In Japan, it can be performed under health insurance.
GCD is a contraindication for LASIK, LASEK, PRK, and SMILE. Postoperatively, corneal opacity rapidly worsens, leading to severe vision loss 1,8,9). After LASIK, numerous small granular deposits form between the flap and the stromal bed. LASIK results in more severe exacerbation and worse final visual acuity compared to PRK 1,8). Case reports from South Korea and Japan describe many patients who were asymptomatic before surgery but developed marked corneal opacity months to years after LASIK, requiring PTK or corneal transplantation 8,9).
QWhat happens if GCD is discovered after undergoing LASIK?
A
In cases of GCD developing after LASIK, granular deposits rapidly form between the flap and the stromal bed. Treatment options include PTK after LASIK flap removal, DALK, and PK, considered in this order. Early consultation with an ophthalmologist is important.
The TGFBI gene encodes TGFBIp (keratoepithelin, 68 kDa), an extracellular matrix protein. TGFBIp is involved in cell adhesion, migration, and proliferation, and is expressed in normal corneal stroma1,5,7). Mutations in the TGFBI gene reduce the susceptibility of mutant TGFBIp to proteolysis, leading to its accumulation as insoluble deposits in the corneal stroma5,7).
GCD2 is almost exclusively caused by the Arg124His (R124H) mutation 1,5). In GCD2, both hyaline and amyloid are deposited.
Impaired autophagy: Impaired autophagy has been reported in GCD2, leading to reduced degradation of TGFBIp and enhanced accumulation 1,5)
Mitochondrial dysfunction: It has been suggested that mutant TGFBIp itself affects corneal fibroblasts and may cause mitochondrial dysfunction 1)
Effect of corneal neovascularization: Deposits tend to decrease and be reabsorbed in areas with corneal neovascularization. This finding supports the mechanism by which deposits concentrate in the avascular central cornea1)
After LASIK, TGFBIp rapidly deposits between the flap and the stromal bed. This is thought to be because surgical manipulation in the central cornea promotes accumulation of mutant TGFBIp1,8). Since corneal incisions during cataract surgery (near the limbus) do not cause exacerbation, it is presumed that the distance from the vascularized limbus is relevant1). Pathological observations by Awwad et al. suggest that deposits formed after LASIK involve TGFBIp accumulation along with keratocyte activation associated with wound healing at the flap-stromal bed interface8).
In GCD1, deposits appear as homogeneous eosinophilic material under light microscopy and stain red with Masson’s trichrome stain. At the electron microscopic level, they are recognized as rod-shaped or trapezoidal electron-dense structures with a diameter of 100–500 nm6).
In GCD2, in addition to hyaline deposits, amyloid fibers (8–10 nm in diameter) are observed. Amyloid fibers stain orange-red with Congo red and show apple-green birefringence under polarized light microscopy6). The dual staining characteristics are useful for definitive pathological diagnosis of GCD2.
According to proteomic analysis by Poulsen et al., mutant R124H-TGFBIp in corneas of GCD2 patients is less susceptible to cleavage by proteolytic enzymes compared to normal TGFBIp, and specific C-terminal fragments selectively accumulate6). This resistance to cleavage is suggested to be the basis for hyaline and amyloid fiber formation.
Lithium chloride has been reported to reduce TGFBI protein production. Combination therapy with melatonin and rapamycin may inhibit TGFBI protein expression while activating autophagy to promote degradation of mutant TGFBIp1,5).
Silencing of mutant TGFBI expression using small interfering RNA (siRNA) or short hairpin siRNA (shRNA) is being studied preclinically. CRISPR/Cas9 genome editing technology is also a candidate, but unintended off-target effects on normal alleles or other genes remain a challenge1,5).
Corneal electrolysis: Experimental use for recurrent cases after corneal transplantation has been reported. Long-term outcomes are unknown.
Machine learning-assisted diagnosis: Development of an AI model that automatically identifies GCD from anterior segment photographs has been reported.
Chaperone therapy: Basic research on chemical chaperones (e.g., 4-phenylbutyric acid) that assist proper folding of mutant TGFBIp is ongoing3).
iPS cell-derived corneal epithelial sheet: Transplantation therapy using corneal epithelial sheets generated from patient-derived iPS cells is being studied as a future option.
In Japan, discussions continue regarding the establishment of a national registry for TGFBI-related corneal dystrophies, led by the Japanese Ophthalmological Society and the Japanese Cornea Society. Since genetic testing was covered by insurance in 2020, the number of genetically diagnosed cases has increased, and long-term follow-up data on mutation patterns and phenotypes specific to the Japanese population are being accumulated3,11). In the future, evidence-based clinical systems are expected to be developed, ranging from carrier screening to determining indications for refractive surgery.
Chang MS, Jun I, Kim EK. Mini-Review: Clinical Features and Management of Granular Corneal Dystrophy Type 2. Korean journal of ophthalmology : KJO. 2023;37(4):340-347. doi:10.3341/kjo.2023.0032. PMID:37336511; PMCID:PMC10427907.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Lisch W, Weiss JS. Clinical and genetic update of corneal dystrophies. Experimental eye research. 2019;186:107715. doi:10.1016/j.exer.2019.107715. PMID:31301286.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern®. Ophthalmology. 2024;131(4):P1-P79.
Poulsen ET, Dyrlund TF, Runager K, et al. Proteomics of granular corneal dystrophy type 2 reveals altered TGFBIp proteolytic processing. J Proteome Res. 2014;13(10):4659-4667.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Awwad ST, Di Pascuale MA, Hogan RN, Forstot SL, McCulley JP, Cavanagh HD. Avellino corneal dystrophy worsening after laser in situ keratomileusis: further clinicopathologic observations and proposed pathogenesis. American journal of ophthalmology. 2008;145(4):656-61. doi:10.1016/j.ajo.2007.12.008. PMID:18243154; PMCID:PMC2661203.
Jun RM, Tchah H, Kim TI, et al. Avellino corneal dystrophy after LASIK. Ophthalmology. 2004;111(3):463-468.
Kitazawa K, Kawasaki S, Shinomiya K, et al. Safety of anterior lamellar keratoplasty for recurrent TGFBI corneal dystrophy. Br J Ophthalmol. 2016;100(4):448-454.