Distrofi kornea granular (GCD) adalah penyakit kornea herediter yang ditandai dengan endapan granular di stroma kornea. Menurut Klasifikasi Internasional Distrofi Kornea edisi kedua (IC3D 2015), termasuk dalam distrofi terkait TGFBI epitel-stroma 2).
Disebabkan oleh mutasi titik pada gen TGFBI (kromosom 5q31) dan diwariskan secara autosomal dominan. Berdasarkan perbedaan mutasi, diklasifikasikan menjadi dua tipe berikut.
Klasifikasi
Mutasi utama
Nama lain/lama
Endapan utama
GCD1
Arg555Trp (R555W)
Granular klasik, Groenouw tipe 1
Hialin saja
GCD2
Arg124His (R124H)
Distrofi Kornea Avellino
Hialin + Amiloid
GCD2 dilaporkan sebagai subtipe independen dari GCD1 pada tahun 1988, dan disebut Distrofi Kornea Avellino karena keluarga pertama berasal dari daerah Avellino, Italia1). Kemudian pada tahun 1997, gen penyebab TGFBI diidentifikasi dan mutasi R124H ditemukan3). Setelah klasifikasi IC3D edisi kedua (2015), secara resmi dinamai Distrofi Kornea Granular tipe 2 (GCD2), dan “Avellino” dicantumkan sebagai nama historis2).
Distrofi kornea granular secara internasional diklasifikasikan dalam kelompok distrofi epitel-stromal. Kelompok ini mencakup enam penyakit terkait TGFBI: granular (GCD1 dan GCD2), kisi (LCD1 dan LCD3A), Reis-Bücklers, dan Thiel-Behnke, semuanya disebabkan oleh mutasi titik berbeda pada gen TGFBI di kromosom 5q312,4). Dalam praktik klinis oftalmologi Jepang, keenam penyakit ini biasa disebut sebagai “Distrofi Kornea terkait TGFBI”.
Distrofi kornea granular pertama kali dilaporkan oleh Groenouw pada tahun 1890, dan saat itu hanya disebut “tipe 1 Groenouw”. Pada tahun 1938, perbedaan dengan distrofi kisi diperjelas, dan untuk waktu yang lama diperlakukan sebagai penyakit tunggal dengan nama “Distrofi Kornea Granular”. Pada tahun 1988, dilaporkan tipe penyakit dengan karakteristik granular dan kisi pada keluarga dari daerah Avellino, Italia, yang kemudian dipisahkan sebagai GCD2 (tipe Avellino)1,2). Revisi klasifikasi IC3D pada tahun 2015 menetapkan sistem klasifikasi saat ini, dan penamaan berdasarkan genotipe menjadi standar internasional2).
Pola pewarisan: Autosomal dominan. Menunjukkan penetrasi tinggi
Tipe 1: Banyak di Eropa dan Amerika. Jarang di Jepang
Tipe 2: Sangat banyak di Asia Timur seperti Jepang dan Korea. Prevalensi di Korea sekitar 11,5 per 10.000 orang1)
Proporsi dalam distrofi kornea terkait TGFBI: GCD2 mencakup 72-91% di Korea dan Jepang, 36% di AS, dan 3% di Polandia1)
Data diagnosis genetik di Jepang: Di Universitas Yamaguchi, 234 pasien distrofi kornea didiagnosis secara genetik selama 21 tahun dari 2000 hingga 2021, dan empat distrofi kornea utama (granular tipe I & II, kisi tipe I & IIIA, tetesan agar-agar, dan makula) mencakup sekitar 96% dari total. - Karakteristik di Asia Timur: Distrofi kornea granular di Asia Timur didominasi tipe 2 (R124H). - Usia onset: Pada heterozigot GCD2, kekeruhan mikroskopis yang hanya terlihat dengan slit lamp dapat diamati sejak usia sekolah, tetapi tidak ada gejala subjektif. Penurunan ketajaman penglihatan subjektif biasanya muncul pada usia 40-50 tahun.
Perbedaan jenis kelamin: Pewarisan autosomal dominan, tidak ada perbedaan jenis kelamin.
QApa yang dimaksud dengan "granular"?
A
Merujuk pada kondisi di mana terdapat banyak gumpalan kecil berwarna putih hingga putih keabu-abuan dengan batas tegas (endapan granular) di lapisan superfisial stroma kornea sentral. Jika diamati langsung dengan slit lamp, digambarkan seperti remah roti, serpihan salju, atau permen konpeito. Endapan berasal dari protein TGFBI mutan.
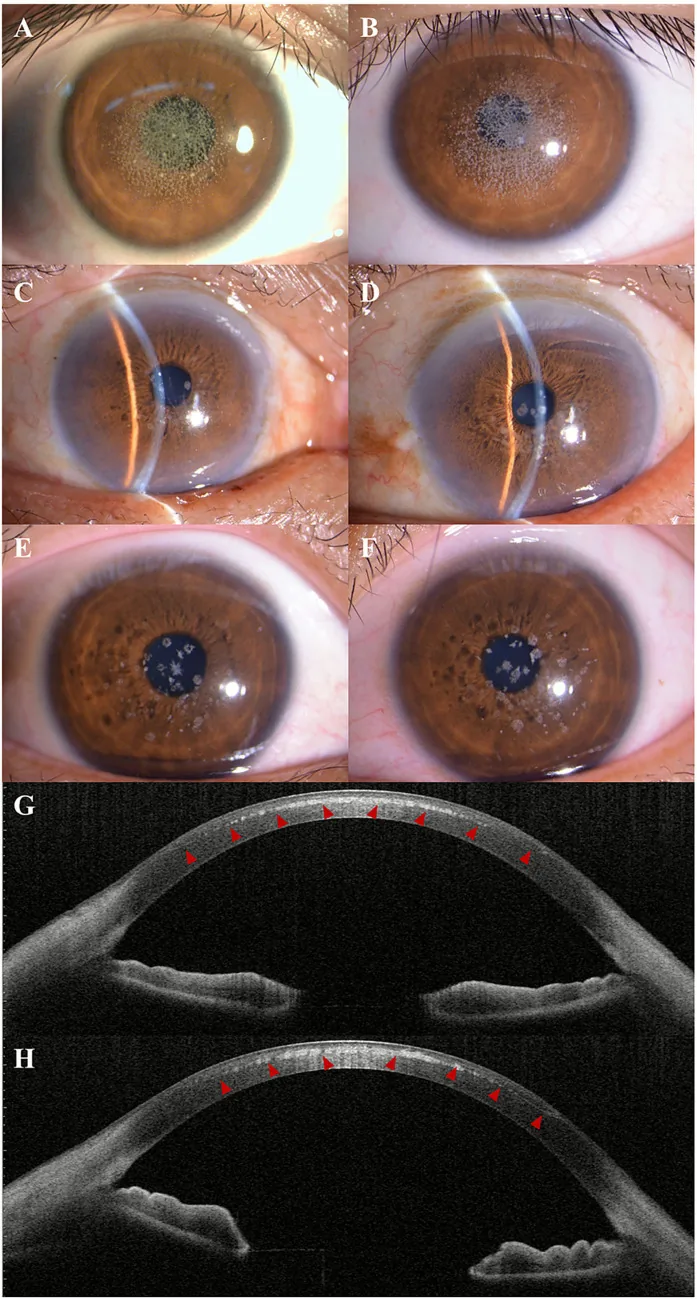
Kuang L, et al. Case Report: Post-LASIK exacerbation of granular corneal dystrophy type 2: a familial case with TGFBI mutation. Front Med (Lausanne). 2025. Figure 1. PMCID: PMC12722859. License: CC BY.
Pada foto slit lamp, tampak kekeruhan granular putih keabu-abuan tersebar dan mengelompok di sentral hingga parasentral kornea. Pada AS-OCT, tampak endapan hiperreflektif di stroma anterior kornea, menunjukkan temuan klinis distrofi kornea granular.
Tanpa gejala hingga ringan: Pada heterozigot, tidak ada penurunan penglihatan yang dirasakan pada fase awal hingga menengah, dan banyak kasus ditemukan secara tidak sengaja saat pemeriksaan. Biasanya, keluhan penurunan penglihatan muncul pada usia 40-50 tahun.
Silau dan fotofobia: Ketika kekeruhan meluas ke area pupil, pasien mengeluh silau di siang hari dan penurunan sensitivitas kontras.
Erosi kornea berulang: Endapan merusak membran Bowman dan membran basal epitel, menyebabkan nyeri tajam, lakrimasi, dan hiperemia saat tidur atau bangun.
Penurunan ketajaman penglihatan: Ketika area transparan di antara endapan menjadi opak, penglihatan menurun secara progresif3).
Kekeruhan granular: Terdapat kekeruhan granular putih hingga putih keabu-abuan yang relatif kecil dengan batas tegas di bagian tengah kornea. Digambarkan seperti remah roti atau serpihan salju.
Kedalaman: Di bawah epitel kornea dan di lapisan superfisial stroma kornea. Tidak meluas ke limbus.
Zat yang mengendap: Hanya hialin. Terwarnai merah dengan pewarnaan Masson’s trichrome. Tidak mengandung amiloid.
Perkembangan: Jumlah granula bertambah seiring bertambahnya usia, dan batas menjadi tidak jelas.
GCD2 (R124H)
Kekeruhan granular: Dimulai dengan kekeruhan putih hingga putih keabu-abuan yang lebih besar dari GCD1 dengan batas tegas. Fenotip bervariasi seperti bentuk gula-gula, linier, bintang, atau seperti gada.
Tipe campuran: Kadang-kadang juga terlihat kekeruhan linier halus seperti jaring yang ditemukan pada distrofi kornea lattice.
Zat yang mengendap: Baik hialin maupun amiloid. Positif pewarnaan Masson’s trichrome dan positif pewarnaan Congo red, menunjukkan warna hijau kekuningan di bawah mikroskop polarisasi.
Perkembangan: Setelah usia 25-30 tahun, ditambahkan kekeruhan putih padat berbentuk batang atau bintang di lapisan tengah stroma. Endapan difus superfisial menjadi lebih parah, menjadikannya indikasi yang baik untuk PTK3).
Pada kedua tipe, kekeruhan terletak di bagian tengah kornea dan tidak meluas ke perifer limbus. Biasanya bilateral dengan sedikit perbedaan antar mata.
Homozigot GCD1: Terdapat kekeruhan retikuler putih pada kedalaman yang sama di bawah epitel kornea hingga stroma superfisial, hampir tanpa celah. Jika progresif, iris dan bilik anterior menjadi tidak dapat diamati.
Homozigot GCD2: Terdapat kekeruhan putih bundar padat di seluruh permukaan kornea kecuali bagian paling perifer, tanpa celah. Cukup parah sehingga warna putih terlihat dengan mata telanjang, hanya transparansi limbus yang dipertahankan 3). Kasus homozigot adalah distrofi kornea refrakter yang kambuh dalam waktu singkat 1-2 tahun setelah PTK atau transplantasi kornea.
QBagaimana perbedaan perjalanan penyakit antara homozigot dan heterozigot?
A
Homozigot muncul pada masa kanak-kanak awal (4-7 tahun) dan progresif cepat. Kekeruhan putih muncul di seluruh kornea tanpa celah, dan PTK atau transplantasi kornea diperlukan sekitar usia 10 tahun. Bahkan setelah operasi, penyakit kambuh dalam 1-2 tahun, menjadikannya perjalanan refrakter. Heterozigot progresif lambat, dan penglihatan baik biasanya dapat dipertahankan hingga usia 40-50 tahun.
GCD disebabkan oleh mutasi titik pada gen TGFBI (kromosom 5q31). Gen TGFBI mengkode protein matriks ekstraseluler TGFBIp (keratoepithelin). TGFBIp mutan memiliki sensitivitas rendah terhadap degradasi protein dan terakumulasi sebagai deposit tidak larut abnormal di stroma kornea1,5,7).
Kelompok distrofi kornea terkait TGFBI meliputi 2,4):
Distrofi kornea granular tipe 1 (R555W)
Distrofi kornea granular tipe 2 (R124H, sebelumnya Avellino)
Distrofi kornea lattice tipe 1 (R124C)
Distrofi kornea lattice tipe 3A (R501T atau L527R)
GCD adalah pewarisan autosomal dominan dengan penetrasi tinggi. Jika probandus telah didiagnosis, 50% kerabat tingkat pertama (orang tua, saudara kandung, anak) mungkin membawa mutasi yang sama. Identifikasi dini pembawa asimtomatik dalam keluarga dapat membantu menghindari operasi refraktif di masa depan dan merencanakan kunjungan rutin untuk pemantauan perkembangan 1,5). Terutama pada individu muda yang menginginkan LASIK, anamnesis riwayat keluarga yang cermat dan tes genetik jika diperlukan sangat dianjurkan.
Diagnosis klinis didasarkan pada pengamatan kekeruhan granular dengan batas tegas di stroma anterior menggunakan slit lamp dan riwayat keluarga positif. Distrofi kornea dicurigai jika ditemukan kekeruhan kornea bilateral (deposit) tanpa kemerahan atau edema kornea3).
Dalam diagnosis banding distrofi kornea, pertama-tama tentukan apakah deposit “berbatas tegas” atau “difus” 3). Jika deposit granular berbatas tegas, bedakan antara GCD1 (kecil) dan GCD2 (besar) berdasarkan ukuran deposit. Pada GCD2, kekeruhan difus seperti lembaran dapat terlihat di antara deposit granular dengan metode sklera scatter, dan kekeruhan difus ini merupakan indikasi yang baik untuk PTK3).
Slit lamp: Pengamatan langsung kekeruhan granular putih dengan batas tegas. Metode sklera scatter, retroluminasi, dan transiluminasi juga digunakan.
OCT segmen anterior (AS-OCT): Menunjukkan kekeruhan reflektif tinggi di stroma anterior. Berguna untuk perencanaan kedalaman eksisi PTK.
Mikroskop konfokal: Menunjukkan kekeruhan tidak teratur dan sangat reflektif seperti remah-remah di antara epitel dan membran Bowman.
Ultrasonografi biomikroskopi (UBM): Mendeteksi granula reflektif tinggi di stroma superfisial.
Analisis topografi kornea: Memberikan informasi tambahan tentang densitas kekeruhan.
Tes genetik: Analisis gen TGFBI berguna untuk diagnosis definitif. Di Jepang, telah dicakup asuransi sejak April 2020 sebagai tes genetik distrofi kornea3).
Fotografi segmen anterior: Penting untuk mengambil foto segmen anterior berkualitas tinggi pada kunjungan pertama dan setiap follow-up untuk dokumentasi observasi jangka panjang.
GCD1: Deposit hialin yang terwarnai merah dengan pewarnaan Masson trichrome. Tidak mengandung amiloid. Di mikroskop elektron, deposit berbentuk batang atau trapesium.
GCD2: Deposit hialin (positif Masson trichrome) dan amiloid (positif Congo red, kuning-hijau di mikroskop polarisasi). Di mikroskop elektron, ditemukan deposit padat elektron berbentuk batang dan fibril amiloid halus 1,7).
Distrofi kornea kisi-kisi tipe 1 (LCD1): Mutasi TGFBI R124C. Kekeruhan linier dan seperti kisi akibat deposisi amiloid di stroma. Sering disertai erosi epitel kornea berulang 3)
Distrofi korneamakula (MCD): Mutasi gen CHST6. Autosomal resesif. Kekeruhan difus di seluruh kornea
Distrofi kornea Reis-Bücklers: Mutasi TGFBI R124L. Kekeruhan seperti peta di lapisan Bowman
Distrofi kornea bercak (FCD): Mutasi PIP5K3. Bintik putih kecil di seluruh stroma, biasanya tanpa gejala
QApakah tes genetik ditanggung asuransi?
A
Sejak April 2020, tes genetik untuk distrofi kornea telah dimasukkan dalam cakupan asuransi. Namun, diperlukan akreditasi fasilitas, sehingga fasilitas yang dapat melakukan tes terbatas. Jika temuan klinis mencurigakan atau jika operasi refraktif seperti LASIK dipertimbangkan, diagnosis pasti melalui tes genetik dianjurkan.
Pada tahap awal tanpa penurunan penglihatan atau erosi epitel berulang, tidak diperlukan pengobatan. Intervensi bedah dipertimbangkan ketika penurunan penglihatan meluas ke area pupil.
Air mata buatan: Gunakan tetes natrium hialuronat 0,1% atau 0,3% 4-6 kali sehari untuk mengurangi kekeringan dan iritasi
Lensa kontak lunak terapeutik: Untuk erosi epitel berulang, melindungi permukaan mata dan mempercepat penyembuhan. Dianjurkan pemakaian sepanjang hari dengan penggantian rutin
Tetes mata dan salep antibiotik: Untuk pencegahan infeksi sekunder selama erosi epitel, gunakan tetes levofloxacin 0,5% 3-4 kali sehari dan salep ofloxacin sebelum tidur
Larutan garam hipertonik (tetes mata atau salep natrium klorida 5%): Kadang digunakan sebagai terapi tambahan untuk mengurangi edema epitel.
Homozigot GCD2: Kambuh sekitar 18 bulan setelah PTK pertama, dan setelah kedua, ketiga, dan seterusnya kambuh dalam waktu sekitar 3 bulan1)
Heterozigot GCD2: Kekambuhan setelah PTK relatif lambat dengan rata-rata 38,4 bulan1)
Penggunaan mitomisin C (MMC) bersamaan: Penggunaan MMC saat PTK tidak dianjurkan. Hal ini karena MMC menginduksi apoptosis keratosit stroma kornea, mengurangi sel yang bertanggung jawab untuk reabsorpsi dan degradasi TGFBIp, dan dapat mempercepat kekambuhan1)
Kasus eksaserbasi pasca LASIK: PTK dapat dilakukan, tetapi efektivitas lebih tinggi setelah pengangkatan flap LASIK1,8)
Perawatan pasca operasi: Setelah PTK, gunakan tetes mata antibiotik (levofloxacin 0,5%) dan tetes mata kortikosteroid (fluorometolon 0,1%) 4 kali sehari hingga epitel sembuh, kemudian kurangi dosis. Penyembuhan epitel biasanya memakan waktu 3-5 hari
DALK adalah teknik yang mempertahankan endotel, dengan mengupas dan mengangkat stroma hingga tepat di atas membran Descemet menggunakan teknik gelembung besar, kemudian menjahit stroma donor. Karena tidak ada risiko penolakan endotel, prognosis jangka panjang dianggap lebih baik daripada PK3). Dalam penelitian Kitazawa dkk., ketajaman visual 5 tahun setelah DALK untuk distrofi kornea terkait TGFBI (termasuk granular dan kisi) umumnya baik, dan tingkat kelangsungan hidup cangkok tinggi10). Di Jepang, prosedur ini dapat dilakukan sebagai perawatan asuransi kesehatan.
GCD merupakan kontraindikasi untuk LASIK, LASEK, PRK, dan SMILE. Setelah operasi, kekeruhan kornea memburuk dengan cepat dan menyebabkan penurunan penglihatan yang berat 1,8,9). Setelah LASIK, terbentuk banyak endapan granular kecil antara flap dan dasar stroma. LASIK menyebabkan perburukan yang lebih parah dibandingkan PRK, dan ketajaman penglihatan akhir juga lebih buruk 1,8). Laporan kasus dari Korea dan Jepang mencatat banyak pasien yang asimtomatik sebelum operasi mengalami kekeruhan kornea yang signifikan dalam beberapa bulan hingga tahun setelah LASIK, sehingga memerlukan PTK atau transplantasi kornea8,9).
QApa yang terjadi jika GCD diketahui setelah menjalani LASIK?
A
Pada kasus GCD yang muncul setelah LASIK, endapan granular terbentuk dengan cepat antara flap dan dasar stroma. Pilihan pengobatan meliputi PTK setelah pengangkatan flap LASIK, DALK, dan PK, yang dipertimbangkan dalam urutan tersebut. Konsultasi dini dengan dokter spesialis mata sangat penting.
Gen TGFBI mengkode protein matriks ekstraseluler TGFBIp (keratoepitelin, 68 kDa). TGFBIp terlibat dalam adhesi sel, migrasi, dan proliferasi, dan diekspresikan di stroma kornea normal 1,5,7). Mutasi pada gen TGFBI menyebabkan TGFBIp mutan menjadi kurang sensitif terhadap degradasi protein, sehingga terakumulasi sebagai endapan tidak larut di stroma kornea5,7).
GCD2 hampir secara eksklusif disebabkan oleh mutasi Arg124His (R124H) 1,5). Pada GCD2, baik hialin maupun amiloid mengendap.
Gangguan Autofagi: Gangguan autofagi telah dilaporkan pada GCD2, yang menyebabkan penurunan degradasi TGFBIp sehingga meningkatkan akumulasi 1,5)
Disfungsi Mitokondria: TGFBIp mutan diduga mempengaruhi fibroblas kornea dan dapat menyebabkan disfungsi mitokondria 1)
Pengaruh neovaskularisasi kornea: Di area yang disertai neovaskularisasi kornea, deposit cenderung berkurang dan diserap kembali. Temuan ini mendukung mekanisme di mana deposit terkonsentrasi di bagian tengah kornea yang tidak memiliki suplai pembuluh darah1)
Setelah LASIK, TGFBIp dengan cepat mengendap di antara flap dan dasar stroma. Hal ini diduga karena manipulasi bedah di bagian tengah kornea mempercepat akumulasi TGFBIp mutan1,8). Karena sayatan kornea pada operasi katarak (dekat limbus) tidak memperburuk kondisi, diperkirakan jarak dari limbus yang memiliki vaskularisasi berperan1). Pengamatan patologis oleh Awwad dkk. menunjukkan bahwa deposit yang terbentuk setelah LASIK terakumulasi bersamaan dengan aktivasi keratosit yang terkait dengan respons penyembuhan luka pada antarmuka flap dan dasar stroma8).
Deposit GCD1 diamati di bawah mikroskop cahaya sebagai substansi asidofilik homogen, dan berwarna merah dengan pewarnaan Masson’s trichrome. Pada tingkat mikroskop elektron, deposit tampak sebagai struktur elektron-densitas tinggi berbentuk batang atau trapesium dengan diameter 100–500 nm6).
Pada GCD2, selain deposit hialin, terdapat serat amiloid (diameter 8–10 nm). Serat amiloid terwarnai oranye-merah dengan pewarnaan Congo red, dan menunjukkan birefringensi hijau apel di bawah mikroskop polarisasi6). Karakteristik pewarnaan ganda ini berguna untuk diagnosis patologis definitif GCD2.
Menurut analisis proteomik oleh Poulsen dkk., TGFBIp mutan R124H pada kornea pasien GCD2 lebih resisten terhadap pemotongan oleh enzim proteolitik dibandingkan TGFBIp normal, dan fragmen terminal-C tertentu terakumulasi secara selektif6). Resistensi pemotongan ini diduga menjadi dasar pembentukan serat hialin dan amiloid.
Lithium klorida dilaporkan mengurangi produksi protein TGFBI. Terapi kombinasi melatonin dan rapamycin menunjukkan kemampuan untuk menghambat ekspresi protein TGFBI sekaligus mengaktifkan autophagy untuk mempercepat degradasi TGFBIp mutan1,5).
Penekanan ekspresi TGFBI mutan menggunakan small interfering RNA (siRNA) atau short hairpin siRNA (shRNA) sedang diteliti secara praklinis. Teknologi pengeditan genom CRISPR/Cas9 juga menjadi kandidat, namun efek off-target yang tidak diinginkan pada alel normal atau gen lain merupakan tantangan1,5).
Elektrolisis kornea (corneal electrolysis): Penggunaan uji coba pada kasus rekuren setelah transplantasi kornea telah dilaporkan. Hasil jangka panjang tidak diketahui.
Dukungan diagnosis dengan pembelajaran mesin: Pengembangan model AI untuk mengidentifikasi GCD secara otomatis dari foto segmen anterior telah dilaporkan.
Terapi chaperone: Penelitian dasar tentang chaperone kimia (misalnya asam 4-fenilbutirat) untuk membantu pelipatan protein TGFBIp mutan sedang berlangsung 3).
Lembaran epitel kornea dari sel iPS: Transplantasi lembaran epitel kornea yang dibuat dari sel iPS pasien sedang diteliti sebagai pilihan masa depan.
Di dalam negeri, diskusi tentang pembentukan registri nasional untuk distrofi kornea terkait TGFBI terus berlanjut, dipimpin oleh Japanese Ophthalmological Society dan Japanese Cornea Society. Sejak cakupan asuransi untuk tes genetik pada tahun 2020, jumlah kasus diagnosis genetik meningkat, dan data tindak lanjut jangka panjang tentang pola mutasi dan fenotipe khas Jepang sedang dikumpulkan 3,11). Ke depannya, diharapkan pengembangan sistem perawatan berbasis bukti mulai dari skrining pembawa hingga penentuan indikasi operasi refraktif.
Chang MS, Jun I, Kim EK. Mini-Review: Clinical Features and Management of Granular Corneal Dystrophy Type 2. Korean journal of ophthalmology : KJO. 2023;37(4):340-347. doi:10.3341/kjo.2023.0032. PMID:37336511; PMCID:PMC10427907.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Lisch W, Weiss JS. Clinical and genetic update of corneal dystrophies. Experimental eye research. 2019;186:107715. doi:10.1016/j.exer.2019.107715. PMID:31301286.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern®. Ophthalmology. 2024;131(4):P1-P79.
Poulsen ET, Dyrlund TF, Runager K, et al. Proteomics of granular corneal dystrophy type 2 reveals altered TGFBIp proteolytic processing. J Proteome Res. 2014;13(10):4659-4667.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Awwad ST, Di Pascuale MA, Hogan RN, Forstot SL, McCulley JP, Cavanagh HD. Avellino corneal dystrophy worsening after laser in situ keratomileusis: further clinicopathologic observations and proposed pathogenesis. American journal of ophthalmology. 2008;145(4):656-61. doi:10.1016/j.ajo.2007.12.008. PMID:18243154; PMCID:PMC2661203.
Jun RM, Tchah H, Kim TI, et al. Avellino corneal dystrophy after LASIK. Ophthalmology. 2004;111(3):463-468.
Kitazawa K, Kawasaki S, Shinomiya K, et al. Safety of anterior lamellar keratoplasty for recurrent TGFBI corneal dystrophy. Br J Ophthalmol. 2016;100(4):448-454.