Distrofi Kornea Tetesan Agar-agar (gelatinous drop-like corneal dystrophy: GDLD) adalah penyakit kornea herediter di mana amiloid mengendap di bawah epitel kornea, menyebabkan penurunan visus yang signifikan pada kedua mata.
Penyakit ini pertama kali dilaporkan pada tahun 1914 oleh Nakaizumi, dan pada tahun 1932 Kiyosawa menamainya “degenerasi kornea tetesan agar-agar”, dan sejak itu disebut demikian. Dalam klasifikasi IC3D (International Committee for Classification of Corneal Dystrophies), diklasifikasikan sebagai distrofi epitel, dengan singkatan GDLD.
Gen penyebab diidentifikasi pada tahun 1999 oleh Tsujikawa dkk., yaitu gen TACSTD2 (tumor-associated calcium signal transducer 2), gen ekson tunggal yang terletak pada kromosom 1p324).
Prevalensi: Jarang secara global, tetapi relatif sering dilaporkan di Jepang 1). Diperkirakan penurunan angka perkawinan sedarah telah menurunkan frekuensi kejadian 2)
Perbedaan regional: Relatif sering ditemukan di Jepang, hampir tidak pernah dilaporkan di Eropa dan Amerika
Mutasi Q118X: Mutasi founder pada pasien Jepang, mencakup lebih dari 80% kromosom penyebab penyakit 2)
Usia onset: Sering timbul sebelum usia 20 tahun
Pada tahun 2019, penyakit ini ditetapkan sebagai penyakit langka spesifik “Distrofi Kornea Tetesan Agar-agar” dan menjadi subjek subsidi biaya pengobatan 2). Kriteria diagnosis dan klasifikasi keparahan telah dibuat oleh Proyek Riset Kebijakan Penyakit Intractable Kementerian Kesehatan, Tenaga Kerja, dan Kesejahteraan 2).
QApakah GDLD juga terjadi di luar Jepang?
A
GDLD telah dilaporkan di seluruh dunia, tetapi lebih sering ditemukan di Jepang. Hampir tidak ada kasus di Eropa dan Amerika. Lebih dari 20 mutasi telah dilaporkan pada gen TACSTD2, menunjukkan heterogenitas genetik. Di Jepang, mutasi Q118X yang terletak pada 1p32 adalah mutasi founder yang sering ditemukan, mencakup lebih dari 80% kromosom penyebab penyakit pada pasien Jepang.
Sering timbul sebelum usia 20 tahun. Pasien mengeluhkan gejala berikut sejak usia dini.
Fotofobia: Gejala yang menonjol sejak tahap awal
Sensasi benda asing: Akibat tonjolan agar-agar pada permukaan kornea
Epifora: Menyertai gejala iritasi
Penurunan visus: Memburuk secara bertahap seiring perkembangan deposit amiloid. Menjadi signifikan setelah dewasa
Seiring bertambahnya usia, jumlah dan ukuran deposit amiloid meningkat. Deposit menjadi putih keabu-abuan hingga kuning, dan akhirnya menutupi sebagian besar kornea di sekitar celah kelopak mata 2). Invasi pembuluh darah dari perifer, penurunan tajam penglihatan, dan nyeri mata terjadi, ditambah masalah kosmetik, yang secara signifikan menurunkan kualitas hidup pasien.
Temuan Klinis (Temuan yang Dikonfirmasi Dokter saat Pemeriksaan)
Kekeruhan kornea diklasifikasikan menjadi 4 tipe berdasarkan morfologi. Tipe-tipe ini dapat dibedakan dengan pemeriksaan segmen anterior menggunakan slit-lamp 2,3).
Seperti Murbei
tipe murbei tipikal: Tipe yang paling khas.
Bagian tengah kornea: Lesi menonjol berwarna putih keabu-abuan yang mengelompok menyerupai murbei.
Amiloid subepitel: Tonjolan koloid semi-transparan berwarna putih susu yang meningkat dari pusat ke perifer.
Tipe Keratopati Pita
tipe keratopati pita: Kadang terlihat pada tahap awal.
Di antara celah kelopak: Kekeruhan lapisan superfisial. Menunjukkan temuan yang mirip dengan keratopati pita.
Lesi konjungtiva: Lesi juga dapat ditemukan di konjungtiva.
Seperti Kumquat
tipe seperti kumquat: Sering pada kasus lanjut.
Deposit kuning-putih difus: Seluruh kornea berubah menjadi kuning dan tampak seperti kumquat.
Invasi pembuluh darah: Dapat disertai neovaskularisasi superfisial.
Tipe kekeruhan stroma
tipe kekeruhan stroma: tahap yang lebih lanjut.
Perluasan ke stroma: Lesi meluas ke stroma kornea.
Invasi pembuluh darah: Disertai invasi pembuluh darah ke dalam tonjolan gelatin berwarna putih susu hingga kuning.
Ide dkk. melaporkan spektrum klinis terperinci dari 34 kasus di Jepang, dan menunjukkan bahwa bahkan dengan mutasi gen TACSTD2 yang sama (homozigot Q118X), empat fenotipe dapat bercampur 3).
Selain itu, terdapat temuan khas berikut:
Pewarnaan fluoresein tertunda: Meskipun tidak ada kelainan epitel kornea, karena peningkatan permeabilitas akibat pembentukan sambungan ketat yang tidak sempurna, fluoresensi diamati beberapa menit setelah pemberian tetes fluoresein2)
Penipisan epitel: Epitel kornea menipis di area dengan tonjolan gelatin
Invasi pembuluh darah: Terdapat invasi pembuluh darah superfisial di perifer kornea
Akibat kelainan gen TACSTD2, lokalisasi intraseluler normal dari Claudin 1 dan Claudin 7, protein penyusun sambungan ketat pada epitel kornea, hilang, sehingga fungsi sawar epitel menurun 5). Akibatnya, protein seperti laktoferin dari air mata masuk ke dalam kornea, membentuk serat amiloid dan mengendap di bawah epitel. Nakatsuka dkk. menunjukkan secara biologi molekuler melalui analisis keluarga Jepang bahwa TACSTD2 penting untuk lokalisasi claudin normal, dan memperjelas bahwa patofisiologi GDLD disebabkan oleh disfungsi sambungan ketat 5).
Riwayat keluarga: Karena merupakan pewarisan resesif autosomal, terdapat risiko terkena jika kedua orang tua adalah pembawa
Keturunan Jepang: Mutasi nonsense Q118X merupakan mutasi pendiri di Jepang, mencakup lebih dari 80% kromosom penyebab penyakit 2)
Perkawinan sedarah: Biasanya orang tua probandus adalah kerabat dekat. Namun, penyakit juga dapat terjadi pada heterozigot majemuk dari perkawinan antar keluarga yang berbeda
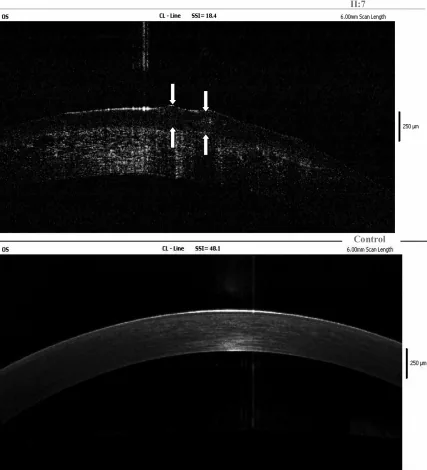
Yang Jing, Chun Liu, Liya Wang A novel TACSTD2 mutation identified in two Chinese brothers with gelatinous drop-like corneal dystrophy 2009 Aug 14 Mol Vis. 2009 Aug 14; 15:1580-1588 Figure 5. PMCID: PMC2728569. License: CC BY.
Temuan OCT domain frekuensi pada kornea kiri probandus menunjukkan deposit amiloid di dalam epitel dan stroma superfisial, dan panah menunjukkan lokasi lesi seperti tetesan agar-agar. Ini sesuai dengan deposit amiloid yang dibahas di bagian “4. Diagnosis dan Metode Pemeriksaan”.
Kriteria Diagnosis (Kelompok Studi Kementerian Kesehatan, Tenaga Kerja, dan Kesejahteraan)
Kriteria diagnosis GDLD telah ditetapkan oleh kelompok studi Proyek Penelitian Kebijakan Penyakit Intractable Kementerian Kesehatan, Tenaga Kerja, dan Kesejahteraan “Pengembangan Pedoman Praktik Klinis untuk Penyakit Intractable Segmen Anterior Mata serta Diseminasi dan Edukasi” 2). Jika diklasifikasikan sebagai Definite berdasarkan kriteria ini, maka menjadi sasaran penyakit langka yang ditetapkan.
A. Gejala (mengalami salah satu)
Penurunan ketajaman penglihatan
Fotofobia
Sensasi benda asing
Air mata berlebih (lakrimasi)
B. Temuan Pemeriksaan
Adanya kumpulan deposit amiloid di bawah epitel kornea (seperti buah murbei) berwarna abu-abu keputihan yang menonjol di bagian tengah kornea hingga celah kelopak pada kedua mata
Adanya pewarnaan tertunda yang terlihat beberapa menit setelah pewarnaan fluoresein meskipun tidak ada kelainan epitel kornea
Adanya invasi pembuluh darah superfisial di perifer kornea
C. Diagnosis banding: Mengeksklusikan amiloidosis kornea sekunder dan keratopati droplet klimatik
D. Komplikasi ekstraokular: Tidak ada
E. Pemeriksaan genetik: Adanya kelainan pada gen TACSTD2
Kondisi Definite terpenuhi jika salah satu dari berikut terpenuhi2).
Kasus yang memenuhi D, salah satu dari A, dan B1, serta dapat mengeksklusikan penyakit yang harus dibedakan pada C
Kasus yang memenuhi D, salah satu dari A, dan B2 atau B3, serta memenuhi E, dan dapat mengeksklusikan penyakit yang harus dibedakan pada C
B1 (deposit seperti murbei) adalah temuan yang sangat khas, dan pada kasus tipikal diagnosis tidak sulit. Pada kasus atipikal, diagnosis dilakukan dengan menggabungkan A-C dan pemeriksaan genetik (E)2).
Klasifikasi keparahan (sesuai pengumuman penyakit langka tertentu)
Keparahan diklasifikasikan ke dalam derajat I-IV berdasarkan ketajaman penglihatan terkoreksi pada mata yang lebih baik2).
Keparahan
Kriteria
Bantuan biaya medis
Derajat I
Hanya satu mata terkena, mata lainnya normal
×
Derajat II
Kedua mata terkena, visus terkoreksi mata terbaik ≥ 0.3
×
Derajat III
Kedua mata terkena, visus terkoreksi mata terbaik ≥ 0.1 dan < 0.3
○
Derajat IV
Kedua mata terkena, visus terkoreksi mata terbaik < 0.1
○
Jika didiagnosis sebagai Definite, maka termasuk penyakit langka yang ditetapkan, dan dapat menerima bantuan biaya medis jika derajat keparahan III atau lebih2). Jika terdapat penyempitan lapang pandang (lapang pandang sentral tersisa ≤ 20 derajat dengan target Goldmann I/4) pada mata yang lebih baik akibat glaukoma sekunder dll., maka derajat keparahan naik satu tingkat.
Pemeriksaan slit-lamp: Amati lesi menonjol keabu-abuan dari pusat kornea hingga area fisura palpebra, dan bedakan 4 tipe (tipe mulberi, tipe pita, tipe kumquat, tipe kekeruhan parenkim)
Uji fluoresensi (pewarnaan tertunda): Karena disfungsi sambungan ketat, pewarna dengan cepat menembus ke dalam jaringan kornea2)
Tes gen TACSTD2: Telah dicakup oleh asuransi kesehatan sejak tahun 2020 sebagai tes gen distrofi kornea (D006-20) 2). TACSTD2 adalah gen ekson tunggal sehingga mudah dicari. Sangat berguna untuk diagnosis kasus atipikal
Pemeriksaan jaringan (irisan kornea): Diwarnai merah jingga dengan pewarnaan Congo red, dan menunjukkan birefringensi hijau apel di bawah mikroskop polarisasi, memastikan amiloid
Amiloidosis kornea sekunder: Amiloid mengendap akibat stimulasi kronis seperti trikiasis, entropion, puncak keratokonus, atau pemakaian lensa kontak keras. Tidak ada riwayat keluarga dan adanya latar belakang inflamasi permukaan mata kronis menjadi titik pembeda. Dapat menunjukkan gambaran seperti gelatin atau kisi-kisi, dan diperlukan pemeriksaan jaringan untuk konfirmasi
Keratopati droplet iklim: Lebih sering pada pria di atas 40 tahun. Ditemukan di daerah gurun atau sangat dingin, disebabkan oleh sinar UV dan kekeringan. Menunjukkan lesi kornea menonjol berwarna kuning hingga putih keabu-abuan
Degenerasi kornea pita: Garam kalsium mengendap di bawah epitel. Dimulai dari perifer pada arah jam 3 dan 9, lalu meluas ke pusat
Distrofi kornea kisi tipe I: Disebabkan mutasi R124C pada gen TGFBI, pewarisan dominan autosomal. Menunjukkan kekeruhan fibrosa bercabang di stroma kornea
Distrofi korneamakula: Disebabkan kelainan gen CHST6, pewarisan resesif autosomal. Kekeruhan difus seperti kaca buram
QApakah tes genetik ditanggung asuransi?
A
Tes gen TACSTD2 telah dicakup oleh asuransi kesehatan sejak tahun 2020 sebagai “Tes gen distrofi kornea (D006-20)”. Namun, fasilitas harus memiliki sistem yang memungkinkan pelaksanaan tes dan mendapatkan akreditasi fasilitas. TACSTD2 adalah gen ekson tunggal sehingga mudah dicari, dan lebih dari 80% pasien Jepang memiliki mutasi pendiri Q118X, sehingga sangat berguna untuk diagnosis kasus atipikal 2).
Pengobatan GDLD dipilih berdasarkan luasnya kekeruhan dan derajat gangguan penglihatan. Karena merupakan penyakit genetik, tantangan terbesar adalah tingkat kekambuhan yang sangat tinggi dengan pengobatan apa pun 2). Tidak jarang komplikasi akibat transplantasi kornea berulang dan glaukoma sekunder menyebabkan kebutaan.
Air mata buatan: Digunakan sebagai terapi simtomatik untuk meredakan gejala iritasi permukaan.
Pemakaian terus-menerus lensa kontak lunak terapeutik (SCL): Dapat menekan kekambuhan lesi gelatinosa dan memperpanjang interval operasi.
Pemakaian terus-menerus SCL terapeutik dipertimbangkan sebagai terapi konservatif dan adjuvant 6). Maeno dkk. pada tahun 2020 dalam studi observasional prospektif pada pasien GDLD menunjukkan bahwa pemakaian SCL terapeutik secara signifikan menekan kekambuhan lesi gelatinosa berwarna abu-abu hingga kuning 7). Juga direkomendasikan untuk pencegahan kekambuhan pasca operasi.
Keratektomi superfisial terapeutik dengan laser eksimer: Pilihan pertama untuk kekeruhan superfisial.
Indikasi: Lesi gelatinosa superfisial derajat ringan hingga sedang. Dikombinasikan dengan kuretase manual. Hasil jangka panjang 8,9).
Transplantasi Kornea
Transplantasi superfisial, deep anterior lamellar (DALK), dan penetrasi penuh (PKP): Diindikasikan untuk kasus lanjut.
Tingkat kekambuhan: Kekambuhan setelah transplantasi kornea penetrasi penuh mencapai 97% dalam 4 tahun. DALK memiliki keuntungan mempertahankan endotel.
Transplantasi Limbus Kornea
Transplantasi sel punca limbal dan pembentukan epitel kornea: Digunakan bersamaan dengan transplantasi kornea.
Tujuan: Menutupi permukaan okular dengan epitel kornea yang berasal dari graft dan mencegah invasi kembali epitel inang 10,11).
Hasil jangka panjang PTK telah dilaporkan dari Jepang. Ōura dkk. menunjukkan hasil jangka panjang PTK pada kasus GDLD dan melaporkan kegunaannya dalam memperpanjang waktu hingga kekambuhan 8). Hieda dkk. dalam studi multisenter Jepang menganalisis secara rinci waktu kekambuhan dan hasil klinis setelah PTK9).
Kombinasi transplantasi sel punca limbal (LSCT) merupakan pendekatan asal Jepang yang diakui secara global. Shimazaki dkk. pada tahun 2002 melaporkan efektivitas transplantasi kornea dengan LSCT untuk GDLD, menunjukkan bahwa hal ini dapat memperpanjang waktu hingga kekambuhan dengan mencegah invasi kembali sel epitel inang 10). Selanjutnya Movahedan dkk. juga melaporkan pendekatan serupa 11).
Untuk menutupi permukaan mata dengan epitel kornea dari cangkok, epitel kornea inang diangkat kemudian dilakukan transplantasi limbus. Pasca operasi, pemakaian lensa kontak terapeutik secara terus-menerus dilanjutkan, dan juga menunda kekambuhan.
Dalam beberapa tahun terakhir, penggunaan kornea buatan (Boston type I Kpro) juga sedang dipertimbangkan. Secara teoritis dapat menghindari pengendapan amiloid berulang karena tidak melalui epitel kornea inang, tetapi ada risiko komplikasi pasca operasi seperti infeksi dan membran retroprostetik.
QApakah akan kambuh meskipun telah menjalani transplantasi kornea?
A
Pada GDLD, tingkat kekambuhan setelah transplantasi kornea sangat tinggi. Dilaporkan sekitar 97% kambuh dalam 4 tahun setelah transplantasi kornea penetrasi (PKP). Penyebab utamanya adalah penggantian sel epitel resipien dengan epitel cangkok. Sebagai tindakan yang berasal dari Jepang, penundaan kekambuhan dilakukan dengan kombinasi transplantasi sel punca limbuskornea10) dan pemakaian lensa kontak terapeutik secara terus-menerus 7). Dalam manajemen jangka panjang, tujuannya adalah mempertahankan fungsi visual dan memperpanjang interval operasi, dengan asumsi kekambuhan.
6. Patofisiologi dan Mekanisme Terjadinya Secara Detail
Gen TACSTD2, gen penyebab GDLD, adalah gen ekson tunggal yang terletak pada kromosom 1p32. Diidentifikasi sebagai gen penyebab pada tahun 1999 oleh Tsujikawa dkk. melalui analisis keterkaitan pada keluarga Jepang 4). Protein TACSTD2 memainkan peran penting dalam mempertahankan fungsi sawar epitel kornea.
Ketika terjadi mutasi kehilangan fungsi pada gen TACSTD2, lokalisasi intraseluler normal dari protein tight junction Claudin 1 dan Claudin 7 terganggu. Nakatsukasa dkk. menunjukkan melalui analisis sel epitel kornea yang dikultur dan keluarga Jepang bahwa kehilangan fungsi TACSTD2 menyebabkan hilangnya lokalisasi Claudin di persimpangan apikolateral dan penurunan fungsi sawar epitel 5). Selanjutnya pada tahun 2011, mereka melaporkan mutasi baru TACSTD2 pada 3 keluarga dan lokalisasi intraselulernya yang abnormal 12).
Karena penurunan fungsi sawar epitel, protein seperti laktoferin dari air mata masuk ke dalam kornea. Laktoferin yang masuk membentuk serat amiloid dan mengendap di bawah epitel kornea. Endapan amiloid mengandung laktoferin, tetapi penyakit ini bukan karena kelainan gen laktoferin.
Secara histologis, kekeruhan seperti susu di bawah epitel menunjukkan warna oranye-merah dengan pewarnaan Congo red, dan menunjukkan birefringensi hijau apel di bawah mikroskop polarisasi. Di bawah mikroskop elektron, tight junction epitel terlihat digantikan oleh ruang elektron-lusen. Endapan juga menembus lamela kornea, menyebabkan degenerasi serat kolagen dan proteoglikan.
Lebih dari 20 mutasi telah dilaporkan pada gen TACSTD212). Di Jepang, mutasi Q118X (mutasi nonsense, fungsional null) merupakan mutasi founder yang mencakup lebih dari 80% kromosom penyebab penyakit2). Penyakit ini biasanya muncul pada homozigot, tetapi juga dapat terjadi pada heterozigot majemuk akibat pernikahan antar keluarga yang berbeda. Menariknya, bahkan pada homozigot Q118X yang sama, keempat subtipe (mulberi, pita, kumquat, dan kekeruhan parenkim) diamati bercampur3).
Amiloidosis kornea diklasifikasikan menjadi primer atau sekunder, sistemik atau lokal. GDLD diklasifikasikan sebagai amiloidosis lokal primer bersama dengan distrofi kornea kisi. Degenerasi amiloid lokal sekunder terjadi akibat trikiasis, keratokonus, trauma, atau pemakaian lensa kontak jangka panjang, dan harus dibedakan secara diagnostik.
GDLD adalah penyakit langka, dan terdapat kekurangan dokter dengan pengalaman klinis di masing-masing fasilitas, serta belum adanya metode diagnosis dan pengobatan standar. Kelompok Penelitian Survei Epidemiologi Penyakit Kornea Langka dan Sulit dari Kementerian Kesehatan, Perburuhan, dan Kesejahteraan, serta kelompok Penelitian Pengembangan dan Diseminasi Pedoman Klinis untuk Penyakit Sulit Segmen Anterior, telah menetapkan kriteria diagnosis dan klasifikasi keparahan2). Pada tahun 2019, “Distrofi Kornea Tetesan Agar-agar” ditetapkan sebagai penyakit langka tertentu, dan saat ini pedoman klinis yang sesuai dengan Minds sedang disusun2).
Dalam jangka panjang, kekambuhan dan perawatan berulang menjadi masalah. Kombinasi lensa kontak terapeutik dan transplantasi limbus dapat menunda kekambuhan dan memperpanjang interval operasi, sehingga meningkatkan prognosis seumur hidup6, 7, 10).
Maeno dkk. melaporkan kasus klinis atipikal yang menunjukkan deposit amiloid berulang hanya pada satu mata, menunjukkan variabilitas fenotipik GDLD13). Pengujian gen TACSTD2 memainkan peran penting dalam diagnosis kasus atipikal tersebut2).
Dalam penelitian dasar, diharapkan adanya penjelasan rinci tentang dinamika molekul tight junction di hilir TACSTD2 dan pengembangan terapi yang menargetkan stabilisasi Claudin. Secara klinis, perluasan indikasi pendekatan regeneratif seperti kornea buatan, transplantasi lembar epitel kornea, dan epitel kornea turunan sel iPS sedang dipertimbangkan.
Ide T, Nishida K, Maeda N, Tsujikawa M, Yamamoto S, Watanabe H, et al. A spectrum of clinical manifestations of gelatinous drop-like corneal dystrophy in japan. American journal of ophthalmology. 2004;137(6):1081-4. doi:10.1016/j.ajo.2004.01.048. PMID:15183793.
Tsujikawa M, Kurahashi H, Tanaka T, Nishida K, Shimomura Y, Tano Y, et al. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nature genetics. 1999;21(4):420-3. doi:10.1038/7759. PMID:10192395.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Tsujikawa M, et al. Tumor-associated calcium signal transducer 2 is required for the proper subcellular localization of claudin 1 and 7: implications in the pathogenesis of gelatinous drop-like corneal dystrophy. Am J Pathol. 2010;177(3):1344-1355. PMID: 20651236. PMCID: PMC2928967. doi:10.2353/ajpath.2010.100149.
Kawasaki S, Kinoshita S. Clinical and basic aspects of gelatinous drop-like corneal dystrophy. Dev Ophthalmol. 2011;48:97-115. PMID: 21540633. doi:10.1159/000324079.
Maeno S, Soma T, Tsujikawa M, Shigeta R, Kawasaki R, Oie Y, et al. Efficacy of therapeutic soft contact lens in the management of gelatinous drop-like corneal dystrophy. The British journal of ophthalmology. 2020;104(2):241-246. doi:10.1136/bjophthalmol-2018-313809. PMID:31023713.
Hieda O, Kawasaki S, Yamamura K, Nakatsukasa M, Kinoshita S, Sotozono C. Clinical outcomes and time to recurrence of phototherapeutic keratectomy in Japan. Medicine. 2019;98(27):e16216. doi:10.1097/MD.0000000000016216. PMID:31277131; PMCID:PMC6635226.
Shimazaki J, Shimmura S, Tsubota K. Limbal stem cell transplantation for the treatment of subepithelial amyloidosis of the cornea (gelatinous drop-like dystrophy). Cornea. 2002;21(2):177-80. doi:10.1097/00003226-200203000-00010. PMID:11862090.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Nishida K, et al. Two novel mutations of TACSTD2 found in three Japanese gelatinous drop-like corneal dystrophy families with their aberrant subcellular localization. Molecular vision. 2011;17:965-70. PMID:21541270; PMCID:PMC3084224.
Maeno S, Soma T, Nishida K. A Case of Clinically Atypical Gelatinous Drop-like Corneal Dystrophy With Unilateral Recurrent Amyloid Depositions. Cornea. 2022;41(11):1447-1450. doi:10.1097/ICO.0000000000003070. PMID:36219213.
Salin teks artikel dan tempelkan ke asisten AI pilihan Anda.
Artikel disalin ke papan klip
Buka asisten AI di bawah, lalu tempelkan teks yang disalin ke kotak chat.