Yang Jing, Chun Liu, Liya Wang A novel TACSTD2 mutation identified in two Chinese brothers with gelatinous drop-like corneal dystrophy 2009 Aug 14 Mol Vis. 2009 Aug 14; 15:1580-1588 Figure 5. PMCID: PMC2728569. License: CC BY.
Ide T, Nishida K, Maeda N, Tsujikawa M, Yamamoto S, Watanabe H, et al. A spectrum of clinical manifestations of gelatinous drop-like corneal dystrophy in japan. American journal of ophthalmology. 2004;137(6):1081-4. doi:10.1016/j.ajo.2004.01.048. PMID:15183793.
Tsujikawa M, Kurahashi H, Tanaka T, Nishida K, Shimomura Y, Tano Y, et al. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nature genetics. 1999;21(4):420-3. doi:10.1038/7759. PMID:10192395.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Tsujikawa M, et al. Tumor-associated calcium signal transducer 2 is required for the proper subcellular localization of claudin 1 and 7: implications in the pathogenesis of gelatinous drop-like corneal dystrophy. Am J Pathol. 2010;177(3):1344-1355. PMID: 20651236. PMCID: PMC2928967. doi:10.2353/ajpath.2010.100149.
Kawasaki S, Kinoshita S. Clinical and basic aspects of gelatinous drop-like corneal dystrophy. Dev Ophthalmol. 2011;48:97-115. PMID: 21540633. doi:10.1159/000324079.
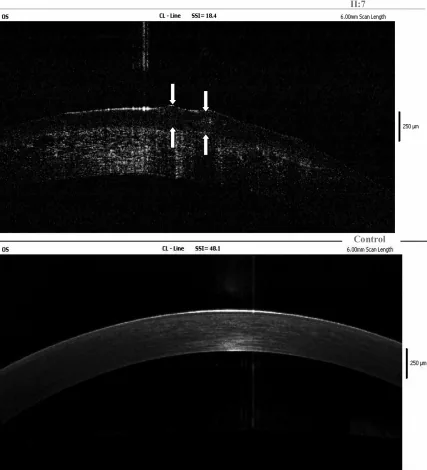
Maeno S, Soma T, Tsujikawa M, Shigeta R, Kawasaki R, Oie Y, et al. Efficacy of therapeutic soft contact lens in the management of gelatinous drop-like corneal dystrophy. The British journal of ophthalmology. 2020;104(2):241-246. doi:10.1136/bjophthalmol-2018-313809. PMID:31023713.
Hieda O, Kawasaki S, Yamamura K, Nakatsukasa M, Kinoshita S, Sotozono C. Clinical outcomes and time to recurrence of phototherapeutic keratectomy in Japan. Medicine. 2019;98(27):e16216. doi:10.1097/MD.0000000000016216. PMID:31277131; PMCID:PMC6635226.
Shimazaki J, Shimmura S, Tsubota K. Limbal stem cell transplantation for the treatment of subepithelial amyloidosis of the cornea (gelatinous drop-like dystrophy). Cornea. 2002;21(2):177-80. doi:10.1097/00003226-200203000-00010. PMID:11862090.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Nishida K, et al. Two novel mutations of TACSTD2 found in three Japanese gelatinous drop-like corneal dystrophy families with their aberrant subcellular localization. Molecular vision. 2011;17:965-70. PMID:21541270; PMCID:PMC3084224.
Maeno S, Soma T, Nishida K. A Case of Clinically Atypical Gelatinous Drop-like Corneal Dystrophy With Unilateral Recurrent Amyloid Depositions. Cornea. 2022;41(11):1447-1450. doi:10.1097/ICO.0000000000003070. PMID:36219213.