La dystrophie cornéenne en gouttes gélatineuses (gelatinous drop-like corneal dystrophy : GDLD) est une maladie cornéenne héréditaire caractérisée par un dépôt d’amyloïde sous l’épithélium cornéen, entraînant une baisse bilatérale marquée de l’acuité visuelle.
Cette maladie a été rapportée pour la première fois en 1914 par Nakaizumi, et depuis que Kiyosawa l’a nommée « dégénérescence cornéenne en gouttes gélatineuses » en 1932, elle est connue sous ce nom. Dans la classification IC3D (International Committee for Classification of Corneal Dystrophies), elle est classée parmi les dystrophies épithéliales, avec l’abréviation GDLD.
Le gène responsable, TACSTD2 (tumor-associated calcium signal transducer 2), a été identifié en 1999 par Tsujikawa et al. Il s’agit d’un gène à exon unique situé sur le chromosome 1p324).
Prévalence : Rare dans le monde, mais rapportée comme relativement fréquente au Japon 1). On pense que la diminution des mariages consanguins a réduit l’incidence 2)
Différence régionale : Relativement fréquente au Japon, rarement rapportée en Europe et en Amérique du Nord
Mutation Q118X : Mutation fondatrice chez les patients japonais, représentant plus de 80 % des chromosomes pathogènes 2)
Âge d’apparition : Survient souvent avant l’âge de 20 ans
En 2019, elle a été désignée comme maladie rare spécifique « Dystrophie cornéenne en gouttes gélatineuses » et est devenue éligible à une aide financière pour les frais médicaux 2). Dans le cadre du projet de recherche sur les politiques relatives aux maladies réfractaires du ministère de la Santé, du Travail et du Bien-être, des critères de diagnostic et une classification de la gravité ont été élaborés 2).
QLa GDLD survient-elle en dehors du Japon ?
A
La GDLD a été rapportée dans le monde entier, mais elle est plus fréquente au Japon. Il n’y a presque aucun cas en Europe et en Amérique du Nord. Plus de 20 mutations du gène TACSTD2 ont été rapportées, montrant une hétérogénéité génétique. Au Japon, la mutation Q118X située en 1p32 est une mutation fondatrice fréquente, représentant plus de 80 % des chromosomes pathogènes chez les patients japonais.
Survient souvent avant l’âge de 20 ans. Les symptômes suivants apparaissent dès l’enfance.
Photophobie : Symptôme marqué dès le début
Sensation de corps étranger : Due aux élévations gélatineuses à la surface de la cornée
Larmoiement : Associé aux symptômes d’irritation
Baisse de l’acuité visuelle : S’aggrave progressivement avec la progression des dépôts d’amyloïde. Devient marquée après l’âge adulte
Avec l’âge, le nombre et la taille des dépôts amyloïdes augmentent. Ils deviennent des dépôts blanc-grisâtre à jaune et finissent par recouvrir la majeure partie de la cornée, en particulier au niveau de la fente palpébrale 2). L’invasion vasculaire à partir de la périphérie, une baisse marquée de l’acuité visuelle et des douleurs oculaires surviennent, et des problèmes esthétiques s’ajoutent, réduisant considérablement la qualité de vie du patient.
Signes cliniques (signes observés par le médecin lors de l’examen)
L’opacité cornéenne est classée en quatre types selon sa morphologie. Ceux-ci peuvent être distingués par l’examen du segment antérieur à l’aide d’un microscope à lampe à fente2,3).
Type mûrier
typical mulberry type : Le type le plus typique.
Partie centrale de la cornée : Des lésions surélevées blanc-grisâtre s’agrègent, ressemblant à une mûre.
Amyloïde sous-épithélial : Des élévations gélatineuses blanc laiteux et translucides augmentent du centre vers la périphérie.
Type kératopathie en bande
band-keratopathy type : Peut être observé dans les premiers stades.
Espace interpalpébral : Opacité superficielle. Présente des similitudes avec la kératopathie en bande.
Lésions conjonctivales : Des lésions peuvent également être observées au niveau de la conjonctive.
Type kumquat
kumquat-like type : Fréquent dans les cas avancés.
Dépôts jaune-blanc diffus : Toute la cornée devient jaune, ressemblant à un kumquat.
Invasion vasculaire : Peut être accompagnée de néovascularisation superficielle.
Type d'opacité stromale
type d’opacité stromale : stade plus avancé.
Extension au stroma : la lésion atteint le stroma cornéen.
Invasion vasculaire : les élévations gélatineuses blanc-jaunâtre s’accompagnent d’une invasion vasculaire.
Ide et al. ont rapporté le spectre clinique détaillé de 34 cas japonais et ont montré que même avec la même mutation du gène TACSTD2 (homozygote Q118X), quatre phénotypes coexistent3).
Les autres signes caractéristiques sont les suivants :
Coloration retardée à la fluorescéine (delayed staining) : malgré l’absence de lésion épithéliale cornéenne, une fluorescence est observée quelques minutes après l’instillation de fluorescéine en raison d’une perméabilité accrue due à une formation incomplète des jonctions serrées2)
Amincissement épithélial : l’épithélium cornéen est aminci au niveau des élévations gélatineuses
Invasion vasculaire : on observe une invasion vasculaire superficielle dans la région périphérique de la cornée
En raison de l’anomalie du gène TACSTD2, la localisation intracellulaire normale de la Claudine 1 et de la Claudine 7, protéines constitutives des jonctions serrées de l’épithélium cornéen, est perdue, ce qui réduit la fonction de barrière épithéliale5). En conséquence, des protéines telles que la lactoferrine des larmes pénètrent dans la cornée, forment des fibrilles amyloïdes et se déposent sous l’épithélium. Nakatsuka et al. ont montré par analyse moléculaire de familles japonaises que TACSTD2 est essentiel à la localisation normale de la claudine, et ont clairement établi que la pathologie de la GDLD est due à un dysfonctionnement des jonctions serrées5).
Antécédents familiaux : en raison de la transmission autosomique récessive, il existe un risque de développer la maladie si les deux parents sont porteurs.
Ascendance japonaise : la mutation non-sens Q118X est une mutation fondatrice au Japon, représentant plus de 80% des chromosomes pathogènes2)
Mariage consanguin : les parents du cas index sont souvent consanguins. Cependant, des hétérozygotes composites issus de mariages entre familles non apparentées peuvent également développer la maladie.
Yang Jing, Chun Liu, Liya Wang A novel TACSTD2 mutation identified in two Chinese brothers with gelatinous drop-like corneal dystrophy 2009 Aug 14 Mol Vis. 2009 Aug 14; 15:1580-1588 Figure 5. PMCID: PMC2728569. License: CC BY.
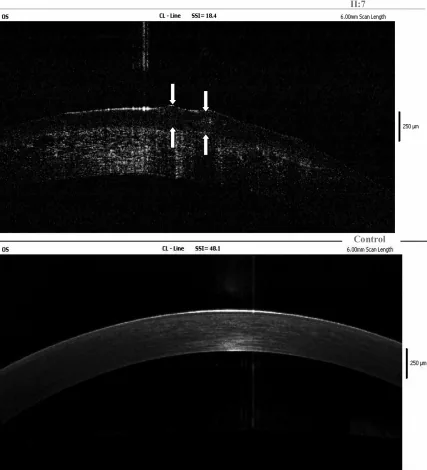
L’examen OCT en domaine de Fourier de l’œil gauche du cas index montre des dépôts d’amyloïde dans l’épithélium cornéen et le stroma superficiel ; les flèches indiquent l’emplacement des lésions gélatineuses en gouttelettes. Cela correspond aux dépôts d’amyloïde traités dans la section « 4. Diagnostic et méthodes d’examen ».
Critères diagnostiques (groupe d’étude du ministère de la Santé)
Les critères diagnostiques de la GDLD ont été établis par le groupe de recherche sur les politiques de santé pour les maladies réfractaires du ministère de la Santé, du Travail et des Affaires sociales, intitulé « Élaboration et diffusion de lignes directrices pour les maladies rares du segment antérieur »2). Un diagnostic « Definite » selon ces critères permet de bénéficier du statut de maladie désignée.
A. Symptômes (au moins un des suivants)
Baisse de l’acuité visuelle
Photophobie
Sensation de corps étranger
Larmoiement
B. Résultats d’examen
On observe une accumulation de dépôts amyloïdes gris-blanc en relief sous l’épithélium cornéen, en forme de mûre, dans la partie centrale de la cornée et la fente palpébrale des deux yeux.
On observe une coloration retardée (delayed staining) : la fluorescence apparaît quelques minutes après la coloration à la fluorescéine, en l’absence de lésion épithéliale cornéenne.
On observe une invasion vasculaire superficielle dans la région périphérique de la cornée.
C. Diagnostic différentiel : exclure l’amylose cornéenne secondaire et la kératopathie climatique en gouttelettes.
Les conditions pour un diagnostic certain sont les suivantes (l’une ou l’autre) 2) :
Cas répondant au critère D, présentant l’un des critères A, remplissant le critère B1, et permettant d’exclure les maladies à différencier en C.
Cas répondant au critère D, présentant l’un des critères A, remplissant le critère B2 ou B3, présentant le critère E, et permettant d’exclure les maladies à différencier en C.
Le critère B1 (dépôts en forme de mûre) est une observation très caractéristique ; dans les cas typiques, le diagnostic n’est pas difficile. Dans les cas atypiques, on combine les critères A à C avec l’examen génétique (E) pour poser le diagnostic 2).
Classification de la sévérité (conformément à la notification des maladies rares désignées)
La sévérité est classée en degrés I à IV en fonction de la meilleure acuité visuelle corrigée de l’œil 2).
Sévérité
Critère
Aide médicale
Grade I
Atteinte d’un seul œil, l’autre œil est sain
×
Grade II
Atteinte des deux yeux, meilleure acuité visuelle corrigée ≥ 0,3
×
Grade III
Atteinte des deux yeux, meilleure acuité visuelle corrigée ≥ 0,1 et < 0,3
○
Grade IV
Atteinte des deux yeux, meilleure acuité visuelle corrigée < 0,1
○
Un diagnostic de Definite permet d’être éligible à la maladie rare désignée, et une aide financière pour les frais médicaux est disponible pour les grades de sévérité III et plus 2). En cas de rétrécissement du champ visuel (champ visuel central résiduel ≤ 20° avec la cible Goldmann I/4) dans le meilleur œil dû à un glaucome secondaire, etc., le grade de sévérité est augmenté d’un niveau.
Examen à la lampe à fente : Observer les lésions surélevées blanc-grisâtre du centre de la cornée à la zone palpébrale, et distinguer les 4 types morphologiques (mûrier, bandelette, kumquat, opacité stromale).
Test de perméabilité à la fluorescéine (coloration retardée) : en raison d’un dysfonctionnement des jonctions serrées, le colorant pénètre rapidement dans le tissu cornéen 2)
Test génétique TACSTD2 : depuis l’année fiscale 2020, il est couvert par l’assurance maladie en tant que test génétique des dystrophies cornéennes (D006-20) 2). TACSTD2 est un gène à exon unique, ce qui facilite la recherche. Particulièrement utile pour le diagnostic des cas atypiques
Examen histologique (coupe de cornée) : la coloration au rouge Congo donne une couleur rouge orangé, et sous microscope polarisant, elle montre une biréfringence vert pomme, confirmant l’amyloïde
Amylose cornéenne secondaire : dépôt d’amyloïde dû à une irritation chronique telle que le trichiasis, l’entropion, la protrusion du cône kératoconique, le port de lentilles de contact rigides. L’absence d’antécédents familiaux et la présence d’une inflammation chronique de la surface oculaire sont des points de différenciation. Peut se présenter sous forme de gélatine ou de motifs en treillis, nécessitant un examen histologique pour confirmation
Kératopathie en gouttelettes climatique : fréquente chez les hommes de plus de 40 ans. Observée dans les régions désertiques ou très froides, due aux UV et à la sécheresse. Présente des lésions cornéennes surélevées jaunes à blanc-gris
Dégénérescence cornéenne en bandelette : dépôt de sels de calcium sous l’épithélium. Commence à la périphérie à 3 et 9 heures et progresse vers le centre
Dystrophie cornéenne grillagée de type I : mutation R124C du gène TGFBI, transmission autosomique dominante. Présente des opacités fibreuses ramifiées dans le stroma cornéen
Dystrophie cornéenne maculaire : anomalie du gène CHST6, transmission autosomique récessive. Opacité diffuse en verre dépoli
QLe test génétique est-il pris en charge par l'assurance maladie ?
A
Le test génétique TACSTD2 est couvert par l’assurance maladie depuis 2020 sous le code « Test génétique des dystrophies cornéennes (D006-20) ». Cependant, l’établissement doit obtenir une certification après avoir mis en place un système permettant d’effectuer le test en interne. TACSTD2 étant un gène à exon unique, facile à rechercher, et plus de 80 % des patients japonais présentant la mutation fondatrice Q118X, ce test est particulièrement utile pour le diagnostic des cas atypiques 2).
Le traitement de la GDLD est choisi en fonction de l’étendue de l’opacité et du degré de déficience visuelle. En raison de la nature héréditaire de la maladie, le taux de récidive est extrêmement élevé quelle que soit la thérapie, ce qui constitue le plus grand défi 2). Les cas de cécité dus à des complications de multiples greffes de cornée ou à un glaucome secondaire ne sont pas rares.
Larmes artificielles : utilisées comme traitement symptomatique pour soulager les symptômes d’irritation de surface.
Port continu de lentilles de contact souples thérapeutiques (SCL) : peut supprimer la récidive des lésions gélatineuses et prolonger l’intervalle entre les interventions chirurgicales.
Le port continu de SCL thérapeutiques est envisagé comme traitement conservateur et adjuvant 6). En 2020, Maeno et al. ont montré dans une étude prospective observationnelle chez des patients atteints de GDLD que le port de SCL thérapeutiques supprimait significativement la récidive des lésions gélatineuses blanc-grisâtre à jaunes 7). Il est également recommandé pour la prévention des récidives postopératoires.
Taux de récidive : Après kératoplastie transfixiante, la récidive est élevée (97 % dans les 4 ans). La DALK présente l’avantage de préserver l’endothélium.
Transplantation limbique
Transplantation de cellules souches limbiques, kératoplastie épithéliale : combinée à la greffe de cornée.
Objectif : Recouvrir la surface oculaire avec l’épithélium cornéen du greffon et empêcher la réinvasion de l’épithélium hôte 10,11).
Plusieurs rapports japonais ont documenté les résultats à long terme de la PTK. Ōura et al. ont montré l’utilité de la PTK pour prolonger l’intervalle avant récidive chez les patients atteints de GDLD8). Hieda et al. ont analysé en détail le moment de la récidive et les résultats cliniques après PTK dans une étude multicentrique japonaise 9).
La combinaison de la transplantation de cellules souches limbiques (LSCT) est une approche d’origine japonaise reconnue mondialement. En 2002, Shimazaki et al. ont rapporté l’efficacité de la greffe de cornée combinée à la LSCT pour la GDLD, montrant qu’elle pouvait prolonger l’intervalle avant récidive en empêchant la réinvasion des cellules épithéliales de l’hôte 10). Par la suite, Movahedan et al. ont également rapporté une approche similaire 11).
Pour recouvrir la surface oculaire avec l’épithélium cornéen du greffon, l’épithélium cornéen de l’hôte est retiré avant de réaliser une greffe de limbe. Après l’opération, le port continu de lentilles de contact thérapeutiques est maintenu pour retarder la récidive.
Ces dernières années, l’indication de la cornée artificielle (Boston Kpro type I) est également envisagée. Théoriquement, elle évite le dépôt d’amyloïde car elle ne passe pas par l’épithélium cornéen de l’hôte, mais il existe des risques de complications postopératoires telles que les infections et la membrane rétroprothétique.
QLa greffe de cornée récidive-t-elle ?
A
Dans la GDLD, le taux de récidive après greffe de cornée est extrêmement élevé. Il a été rapporté qu’environ 97 % des cas récidivent dans les 4 ans suivant une kératoplastie transfixiante (PKP). La cause principale est le remplacement de l’épithélium du greffon par les cellules épithéliales du receveur. Comme mesures développées au Japon, l’association d’une greffe de cellules souches limbiques cornéennes 10) et le port continu de lentilles de contact thérapeutiques 7) visent à retarder la récidive. Dans la gestion à long terme, l’objectif est de maintenir la fonction visuelle et d’allonger l’intervalle entre les interventions, en partant du principe que la récidive surviendra.
6. Physiopathologie et mécanisme détaillé de la maladie
Le gène TACSTD2, responsable de la GDLD, est un gène à exon unique situé sur le chromosome 1p32. En 1999, Tsujikawa et al. l’ont identifié comme gène responsable par analyse de liaison dans des familles japonaises 4). La protéine TACSTD2 joue un rôle essentiel dans le maintien de la fonction de barrière de l’épithélium cornéen.
Lorsqu’une mutation de perte de fonction survient dans le gène TACSTD2, la localisation intracellulaire normale des protéines de jonction serrée Claudine 1 et Claudine 7 est perturbée. Nakatsuka et al. ont montré, par analyse de cellules épithéliales cornéennes en culture et de familles japonaises, que la perte de fonction de TACSTD2 entraîne une perte de localisation des claudines à la jonction apicolatérale et une diminution de la fonction de barrière épithéliale 5). En 2011, ils ont également rapporté de nouvelles mutations de TACSTD2 dans trois familles et leur localisation intracellulaire anormale 12).
En raison de la diminution de la fonction de barrière épithéliale, des protéines telles que la lactoferrine des larmes pénètrent dans la cornée. La lactoferrine ainsi pénétrée forme des fibrilles amyloïdes qui se déposent sous l’épithélium cornéen. Les dépôts amyloïdes contiennent de la lactoferrine, mais cette maladie n’est pas due à une anomalie du gène de la lactoferrine.
Histologiquement, l’opacité laiteuse sous-épithéliale se colore en orange-rouge avec le rouge Congo et présente une biréfringence vert pomme en microscopie à lumière polarisée. En microscopie électronique, on observe que les jonctions serrées de l’épithélium sont remplacées par des espaces clairs aux électrons. Les dépôts envahissent également les lamelles cornéennes, entraînant une dégénérescence des fibres de collagène et des protéoglycanes.
Plus de 20 mutations du gène TACSTD2 ont été rapportées 12). Au Japon, la mutation Q118X (mutation non-sens, fonctionnelle nulle) est une mutation fondatrice représentant plus de 80 % des chromosomes pathogènes 2). La maladie survient typiquement chez les homozygotes, mais elle peut également se développer chez des hétérozygotes composites issus de mariages entre familles différentes. Fait intéressant, même chez les homozygotes Q118X, les quatre formes cliniques (mûrier, en bande, kumquat, et opacité stromale) sont observées de manière mixte 3).
Les amyloïdoses cornéennes sont classées en primaires ou secondaires, systémiques ou locales. La GDLD, avec la dystrophie cornéenne grillagée, est classée parmi les amyloïdoses primaires locales. La dégénérescence amyloïde secondaire locale survient en association avec le trichiasis, le kératocône, les traumatismes, le port prolongé de lentilles de contact, etc., et doit être envisagée dans le diagnostic différentiel.
La GDLD est une maladie rare, et il y avait un problème de manque de médecins expérimentés dans chaque établissement, ainsi que l’absence de méthodes standardisées de diagnostic et de traitement. Les critères diagnostiques et la classification de sévérité ont été élaborés par le groupe de recherche sur l’épidémiologie des maladies cornéennes rares et réfractaires du ministère de la Santé, du Travail et des Affaires sociales, et par le groupe de recherche sur l’élaboration et la diffusion de directives cliniques pour les maladies rares du segment antérieur 2). En 2019, la GDLD a été reconnue comme maladie rare désignée sous le nom de « dystrophie cornéenne en gouttes gélatineuses », et des directives cliniques conformes à Minds (Medical Information Network Distribution Service) sont en cours d’élaboration 2).
À long terme, les récidives et les traitements multiples posent problème. L’utilisation combinée de lentilles de contact thérapeutiques et de greffe de limbe peut retarder la récidive et prolonger l’intervalle entre les interventions chirurgicales, améliorant ainsi le pronostic à vie 6, 7, 10).
Rapports de cas atypiques et de récidive unilatérale
Maeno et al. ont rapporté un cas clinique atypique montrant un dépôt amyloïde récurrent uniquement dans un œil, démontrant la diversité phénotypique de la GDLD13). Le test génétique du gène TACSTD2 joue un rôle déterminant dans le diagnostic de ces cas atypiques 2).
En recherche fondamentale, on espère élucider en détail la dynamique des molécules de jonction serrée en aval de TACSTD2 et développer des thérapies ciblant la stabilisation de la claudine. Sur le plan clinique, l’extension des indications des approches de médecine régénérative, telles que les cornées artificielles, les greffes de feuillets épithéliaux cornéens et les épithéliums cornéens dérivés de cellules iPS, est à l’étude.
Ide T, Nishida K, Maeda N, Tsujikawa M, Yamamoto S, Watanabe H, et al. A spectrum of clinical manifestations of gelatinous drop-like corneal dystrophy in japan. American journal of ophthalmology. 2004;137(6):1081-4. doi:10.1016/j.ajo.2004.01.048. PMID:15183793.
Tsujikawa M, Kurahashi H, Tanaka T, Nishida K, Shimomura Y, Tano Y, et al. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nature genetics. 1999;21(4):420-3. doi:10.1038/7759. PMID:10192395.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Tsujikawa M, et al. Tumor-associated calcium signal transducer 2 is required for the proper subcellular localization of claudin 1 and 7: implications in the pathogenesis of gelatinous drop-like corneal dystrophy. Am J Pathol. 2010;177(3):1344-1355. PMID: 20651236. PMCID: PMC2928967. doi:10.2353/ajpath.2010.100149.
Kawasaki S, Kinoshita S. Clinical and basic aspects of gelatinous drop-like corneal dystrophy. Dev Ophthalmol. 2011;48:97-115. PMID: 21540633. doi:10.1159/000324079.
Maeno S, Soma T, Tsujikawa M, Shigeta R, Kawasaki R, Oie Y, et al. Efficacy of therapeutic soft contact lens in the management of gelatinous drop-like corneal dystrophy. The British journal of ophthalmology. 2020;104(2):241-246. doi:10.1136/bjophthalmol-2018-313809. PMID:31023713.
Hieda O, Kawasaki S, Yamamura K, Nakatsukasa M, Kinoshita S, Sotozono C. Clinical outcomes and time to recurrence of phototherapeutic keratectomy in Japan. Medicine. 2019;98(27):e16216. doi:10.1097/MD.0000000000016216. PMID:31277131; PMCID:PMC6635226.
Shimazaki J, Shimmura S, Tsubota K. Limbal stem cell transplantation for the treatment of subepithelial amyloidosis of the cornea (gelatinous drop-like dystrophy). Cornea. 2002;21(2):177-80. doi:10.1097/00003226-200203000-00010. PMID:11862090.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Nishida K, et al. Two novel mutations of TACSTD2 found in three Japanese gelatinous drop-like corneal dystrophy families with their aberrant subcellular localization. Molecular vision. 2011;17:965-70. PMID:21541270; PMCID:PMC3084224.
Maeno S, Soma T, Nishida K. A Case of Clinically Atypical Gelatinous Drop-like Corneal Dystrophy With Unilateral Recurrent Amyloid Depositions. Cornea. 2022;41(11):1447-1450. doi:10.1097/ICO.0000000000003070. PMID:36219213.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.