La dystrophie cornéenne en treillis (lattice corneal dystrophy, LCD) est une dystrophie cornéenne héréditaire caractérisée par un dépôt d’amyloïde dans le stroma cornéen, produisant des opacités linéaires en treillis. Décrite dès les années 1890, elle est classée dans la 2e édition de l’IC3D (International Committee for Classification of Corneal Dystrophies) en LCD1 et ses variants (anciens types 3, 3A, 1/3A, 4)4).
Le LCD1, la dystrophie cornéenne granuleuse, la dystrophie cornéenne de Reis-Bücklers et la dystrophie cornéenne de Thiel-Behnke forment un groupe de maladies appelées « dystrophies liées au TGFBI ». Le gène responsable TGFBI (transforming growth factor beta-induced gene) est situé sur le bras long du chromosome 5 (5q31) et suit un mode de transmission autosomique dominant. La protéine TGFBI (TGFBIp, kerato-épithéline, βig-h3) est produite par l’épithélium cornéen et distribuée dans toutes les couches de la cornée. Dans le stroma cornéen, elle participe à l’organisation des fibres de collagène. Même pour des mutations du même gène, la différence de site de mutation ou de substitution d’acides aminés entraîne une grande diversité dans la substance déposée (hyaline ou amyloïde) et le tableau clinique5).
La mutation représentative de la LCD1 est R124C, où l’arginine en position 124 du gène TGFBI est remplacée par une cystéine. Dans la variante LCD IIIA, des mutations telles que L527R ont été rapportées.
Diagnostic tissulaire et caractéristiques classiques
La protéine anormale accumulée dans la cornée se colore en rouge au rouge Congo et présente une biréfringence vert pomme caractéristique en microscopie à lumière polarisée, confirmant ainsi la présence d’amyloïde. Cette observation est un indicateur diagnostique classique de l’amylose depuis le 19e siècle6).
Position de l’ancienne LCD2 (syndrome de Meretoja)
Le type anciennement appelé « dystrophie cornéenne grillagée de type 2 » est une manifestation oculaire de l’amylose systémique à gelsoline (GSN-AMYL, syndrome de Meretoja). Dans la classification IC3D actuelle, il est classé comme « amylose familiale » et traité indépendamment de la LCD classique4,10). Ce syndrome, décrit par Meretoja en Finlande en 1969, est une maladie héréditaire associant des opacités cornéennes grillagées, une neuropathie crânienne progressive, un relâchement cutané et des symptômes systémiques10,11). Comme leur distinction est importante en pratique clinique, cet article présente les deux entités.
Opacités filamenteuses à double contour dans la zone pupillaire, érosions épithéliales récurrentes
LCD IIIA (type variant)
TGFBI (5q31)
L527R, etc.
Après 40 ans
Lignes réticulaires épaisses en corde dans le stroma profond, pas d’atteinte épithéliale
Type GSN (Meretoja)
GSN (9q34)
D187N, p.Glu580Lys2)
30-40 ans
Lignes réticulaires radiales en périphérie, amylose systémique
Au Japon, la dystrophie cornéenne liée à TGFBI la plus fréquente est le type granulaire II (type Avellino, R124H), tandis que la LCD1 est moins courante. Cependant, comme les deux types diffèrent par seulement quelques bases du même gène TGFBI, un test génétique est souhaitable pour confirmer le diagnostic dans les cas où les tableaux cliniques se chevauchent. La prévalence exacte de l’ensemble des LCD au Japon n’a pas été rapportée, mais elles sont relativement rares parmi les dystrophies cornéennes.
QQuelle est la différence entre la LCD1 et le syndrome de Meretoja ?
A
La LCD1 est un dépôt amyloïde limité à la cornée dû à une mutation du gène TGFBI, débutant dans la zone pupillaire entre 10 et 20 ans et fréquemment associé à des érosions épithéliales récurrentes. En revanche, le syndrome de Meretoja (anciennement LCD2, type GSN) est une manifestation oculaire d’une amylose systémique due à une mutation du gène GSN (gelsoline), débutant en périphérie cornéenne entre 30 et 40 ans, avec une transparence centrale longtemps préservée. Le syndrome de Meretoja s’accompagne de symptômes systémiques tels qu’un relâchement cutané, un faciès masqué, une neuropathie périphérique et des arythmies cardiaques2,10). Dans la 2e édition de l’IC3D, le syndrome de Meretoja est classé indépendamment de la dystrophie cornéenne réticulée4).
Liu Z, et al. An R124C mutation in TGFBI caused lattice corneal dystrophy type I with a variable phenotype in three Chinese families. Mol Vis. 2008. Figure 2. PMCID: PMC2443752. License: CC BY.
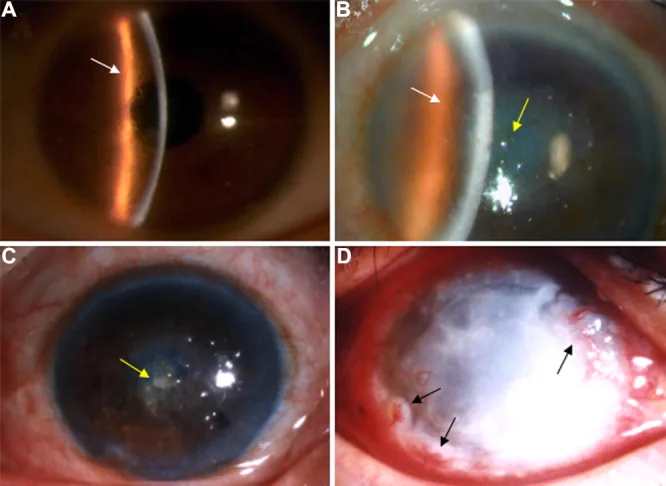
Sur la photographie à la lampe à fente, on observe des lignes réticulées ramifiées dans le stroma cornéen et une opacité prédominante au centre. Cette image montre les signes cliniques typiques de la dystrophie cornéenne grillagée.
Dans le LCD1, la plupart des patients sont asymptomatiques pendant l’enfance, avec seulement de fines opacités détectables par rétroéclairage à la lampe à fente. À partir de la deuxième ou troisième décennie, des érosions cornéennes récurrentes (RCE) se produisent, provoquant des douleurs oculaires intenses, une photophobie, un larmoiement et une sensation de corps étranger au réveil. Vers l’âge de 30 ans, une opacité blanche apparaît dans le stroma antérieur central, et la vision diminue progressivement après 40 ans.
Dans le LCD IIIA (type variant), les lésions épithéliales ne surviennent généralement pas, et le principal symptôme est une baisse lente de la vision après 40 ans.
Dans l’ancien LCD2 (syndrome de Meretoja), les symptômes oculaires apparaissent entre 30 et 40 ans, mais une déficience visuelle significative est souvent retardée jusqu’à la soixantaine 11). Des symptômes systémiques tels qu’un relâchement cutané palpébral, un faciès masqué, une neuropathie crânienne progressive et des arythmies cardiaques précèdent ou accompagnent fréquemment les symptômes oculaires 2,10).
Les résultats à la lampe à fente pour chaque type sont présentés ci-dessous.
LCD1 (type classique)
Site initial : Apparaît dans la zone pupillaire des deux yeux sous forme de fines opacités ponctuées et linéaires dans la couche de Bowman et le stroma antérieur.
Lignes grillagées : Des opacités filamenteuses et linéaires à double contour s’entrelacent pour former des opacités réticulées et étoilées.
Stade avancé : Une opacité blanc laiteux de forme ovale ou circulaire apparaît dans la cornée centrale.
Rétroéclairage : Les fines lignes grillagées translucides, difficiles à voir en illumination directe, deviennent clairement visibles.
Coloration à la fluorescéine : En raison de la diminution de l’adhésion épithéliale, la surface devient rugueuse.
Érosions épithéliales récurrentes : Fréquentes car les dépôts s’étendent aux cellules basales de l’épithélium et à la membrane de Bowman.
LCD IIIA (type variant)
Lignes grillagées : lignes grillagées épaisses et longues dans le stroma moyen à profond, parfois avec des ramifications dendritiques. Observables même en éclairage direct.
Phénotype : trois motifs : ① lignes grillagées uniquement, ② petites déposits granuleux uniquement, ③ mélange des deux. Il peut y avoir des phénotypes différents entre les deux yeux d’un même individu ou des cas unilatéraux.
Épithélium : généralement, aucun trouble épithélial ne se produit.
Homozygotes : chez les homozygotes L527R, les lignes grillagées sont plus épaisses et les dépôts granuleux centraux sont plus gros, mais la différence n’est pas aussi marquée qu’entre hétéro- et homozygotes pour R124H (type granuleux II).
Type GSN (Meretoja)
Lignes grillagées : quelques dépôts grillagés peu fins apparaissent radialement à partir de la périphérie.
Transparence centrale : la transparence centrale est maintenue longtemps après le début.
Érosion épithéliale : rare.
Manifestations systémiques : changements faciaux tels que faciès figé, lèvres proéminentes avec troubles moteurs, oreilles tombantes, blépharochalasis 2).
Dans LCD1, certains cas présentent une opacité circulaire centrale particulièrement prononcée ; un rapport décrit un hétérozygote R124C de 56 ans ayant nécessité une greffe de cornée en raison d’une opacité circulaire centrale.
QPeut-on diagnostiquer LCD1 chez un enfant ?
A
La plupart des LCD1 chez l’enfant sont asymptomatiques et difficiles à détecter en éclairage direct seul. Un examen détaillé à la lampe à fente en transillumination ou en rétro-illumination permet de confirmer de fines opacités ponctuelles à linéaires dans le stroma superficiel central. Chez les enfants présentant des érosions épithéliales récurrentes, il est recommandé de suspecter LCD1, de recueillir les antécédents familiaux et d’examiner les cornées des parents. Le test génétique TGFBI est utile pour le diagnostic définitif.
Les gènes responsables et les mutations représentatives de la dystrophie cornéenne grillagée sont résumés ci-dessous.
Lié à TGFBI (LCD1, LCD IIIA, LCD IV)
Locus génétique : 5q31 (gène TGFBI).
Mode de transmission : Autosomique dominant.
Mutation représentative du LCD1 : R124C (Arg124Cys) est la plus fréquente5).
Mutation représentative du LCD IIIA : L527R (Leu527Arg) a été rapportée. Des cas homozygotes existent également.
Mutation de novo : Une mutation de novo L509P du gène TGFBI a été rapportée dans un cas présentant le phénotype LCD IIIA1). Les parents n’avaient pas la mutation, mais elle a été transmise à un enfant1).
Rôle de la TGFBIp : Produite par l’épithélium cornéen, elle est distribuée dans toute l’épaisseur de la cornée et participe à l’organisation des fibres de collagène dans le stroma5).
Lié à GSN (syndrome de Meretoja, anciennement LCD2)
Locus génétique : 9q34 (gène GSN, gelsoline).
Mode de transmission : Autosomique dominant.
Mutation classique : D187N (type finlandais) est la plus fréquente ; p.Asp187Tyr a également été rapportée10,11).
Nouvelle mutation : p.Glu580Lys, rapportée dans une famille slovène, est située à la frontière des domaines G4-G5 et provoque une répulsion électrostatique par substitution d’une charge négative par une charge positive2).
Tableau clinique : En plus des opacités cornéennes en treillis, se manifeste une amylose systémique avec relâchement cutané, arythmie cardiaque, atteinte rénale et neuropathie optique2).
Étant une maladie héréditaire, les antécédents familiaux constituent le facteur de risque le plus important. Cependant, des mutations de novo du gène TGFBI peuvent survenir, donc l’absence d’antécédents familiaux ne permet pas d’exclure la maladie1). La transmission est autosomique dominante : si l’un des parents est porteur de la mutation, le risque de transmission à l’enfant est de 50 %. Il n’y a pas de différence entre les sexes, et les différences raciales ne sont pas évidentes pour le LCD1, mais le syndrome de Meretoja est connu pour sa prévalence en Finlande avec des familles nombreuses11).
La contribution des facteurs environnementaux n’est pas claire ; l’apparition et la progression de cette maladie sont principalement déterminées par le génotype. Cependant, la fréquence des érosions épithéliales récurrentes peut être aggravée par un environnement sec, le port de lentilles de contact ou un traumatisme. La chirurgie réfractive (LASIK, SMILE, etc.) peut provoquer une aggravation rapide des dystrophies cornéennes liées au TGFBI ; une attention particulière est nécessaire lors du dépistage préopératoire des cas avec antécédents familiaux5).
Pour différencier la LCD1, les formes variantes et le type GSN, il faut combiner les résultats de la lampe à fente, de l’histologie et de la génétique.
Examens cliniques
Microscope à lampe à fente : En éclairage direct, les lignes grillagées précoces sont facilement négligées. En transillumination, on détecte de fines opacités sur fond pupillaire, et en rétro-éclairage, de fines lignes grillagées translucides.
Coloration à la fluorescéine : Dans la LCD1, la diminution de l’adhésion épithéliale rend la coloration « rugueuse ». Elle est également utile pour évaluer l’étendue de l’érosion épithéliale.
Microscopie confocale cornéenne : Elle permet d’observer les dépôts dans le stroma au niveau cellulaire.
Diagnostic de certitude
Test génétique : La détection de mutations dans les gènes TGFBI et GSN permet de confirmer le type de la maladie. Même pour un même phénotype, des mutations différentes entraînent des vitesses de récidive et de progression différentes, ce qui a un impact direct sur le plan de traitement.
Examen histopathologique : La coloration au rouge Congo donne une couleur rouge et montre une biréfringence vert pomme en lumière polarisée, confirmant ainsi la présence d’amyloïde6).
Immunohistochimie : La différenciation des types de la maladie est possible à l’aide d’anticorps anti-TGFBIp et anti-gelsoline.
Recueil des antécédents familiaux : En raison de la transmission autosomique dominante, la confirmation des résultats cornéens chez les parents et la fratrie soutient le diagnostic.
Dystrophie cornéenne granuleuse de type II (type Avellino, TGFBI R124H) : C’est la dystrophie liée à TGFBI la plus fréquente au Japon, montrant un mélange de dépôts granuleux et de lignes grillagées. Pour la différencier de la LCD1, le test génétique est fiable.
Amylose cornéenne secondaire : non héréditaire, l’amylose se dépose secondairement dans un contexte de stimulation chronique de la surface oculaire comme le trichiasis ou le kératocône. L’absence d’antécédents familiaux et la présence d’une maladie sous-jacente sont des points de différenciation.
Dystrophie maculaire de la cornée : hérédité autosomique récessive due à une anomalie du gène CHST6, avec opacité diffuse en verre dépoli et anomalies endothéliales.
Dystrophie cornéenne en gouttes gélatineuses : hérédité autosomique récessive due à une anomalie du gène TACSTD2, se présentant sous forme de saillies gélatineuses blanc laiteux. Relativement fréquente au Japon.
QPourquoi les tests génétiques sont-ils importants ?
A
Dans la dystrophie cornéenne grillagée, même si le phénotype est similaire, des différences dans le gène causal et le site de mutation entraînent des variations importantes dans la vitesse de progression, la fréquence des récidives, les options thérapeutiques et la présence de complications systémiques. La LCD1 due à une mutation TGFBI et le syndrome de Meretoja dû à une mutation GSN diffèrent fondamentalement en termes de stratégie thérapeutique et de nécessité d’un bilan systémique 2,10). De plus, des cas de mutation de novo ont été rapportés, où les antécédents familiaux seuls ne permettent pas de déterminer le type de maladie 1), ce qui rend les tests génétiques indispensables pour le diagnostic définitif et la classification.
Chez l’enfant et l’adulte jeune, en l’absence de symptômes ou en présence d’opacités fines uniquement, une surveillance est recommandée. L’évolution est évaluée par un examen à la lampe à fente tous les six mois à un an.
Prise en charge des érosions épithéliales récurrentes
Pour les érosions épithéliales récurrentes, symptôme central de la LCD1, le traitement conservateur suivant constitue la première étape :
Traitement des crises : port continu de lentilles de contact souples thérapeutiques pour protéger l’épithélium cornéen. Association de collyres antibiotiques pour prévenir une surinfection. Application de pommade ophtalmique pour lubrifier et protéger l’épithélium.
Prévention des récidives : l’administration de pommade ophtalmique avant le coucher réduit le risque de récidive des crises d’érosion épithéliale récurrente. En environnement sec, utiliser des larmes artificielles ou des lubrifiants également pendant la journée.
Dans la LCD1, où les dépôts amyloïdes sont principalement superficiels, en cas d’opacité centrale importante ou d’érosions épithéliales récurrentes fréquentes, la photokératectomie thérapeutique (PTK) au laser excimer est le traitement de première intention 7,8). En général, une récidive précoce ne se produit pas, mais une récidive à long terme est inévitable ; le traitement PTK peut être effectué jusqu’à deux fois sur le même œil.
Chez les hétérozygotes, la récidive est lente et les cas nécessitant un retraitement sont rares. Chez les homozygotes, la récidive a tendance à survenir plus tôt que chez les hétérozygotes. Comme pour les autres dystrophies liées à TGFBI, le taux de récidive après PTK augmente avec le temps, et un suivi à long terme révèle des signes de récidive dans la plupart des cas8).
À titre d’exemple de l’efficacité de la PTK, dans un cas de LCD IIIA dû à une mutation de novo TGFBI L509P, une PTK de 60 µm guidée par FD-OCT a amélioré la meilleure acuité visuelle corrigée (MAVC) de 20/400 à 20/501). Aucune diminution de l’acuité visuelle ni récidive significative n’a été observée à 45 mois postopératoires1).
Selon le Preferred Practice Pattern de l’AAO sur l’œdème et l’opacité cornéens, la PTK pour les dystrophies cornéennes granuleuses et grillagées est une « option raisonnable » et peut retarder le recours à la DALK ou à la greffe de cornée totale, mais comporte un risque de haze postopératoire. En cas de répétition, l’utilisation concomitante de mitomycine C est envisagée pour supprimer les cicatrices récurrentes et les dépôts stromaux. Il est averti que le risque d’ectasie cornéenne augmente si l’ablation dépasse le tiers antérieur du stroma ou si le lit résiduel est inférieur à 250 µm7).
En cas de récidives répétées ou d’opacité s’étendant au-delà du stroma moyen, on opte pour une greffe de cornée. Dans la LCD1, la greffe n’est généralement pas indiquée avant l’âge de 40 ans. Comme les cellules endothéliales sont normalement intactes dans la LCD, la technique chirurgicale est choisie en fonction de la profondeur de l’opacité.
Bonne récupération visuelle mais risque de rejet et de récidive
Ces dernières années, en raison de la réduction du risque de rejet et de résultats visuels comparables à ceux de la kératoplastie transfixiante, la DALK est devenue largement utilisée comme nouveau traitement de première intention.
La récidive de la LCD après une greffe de cornée est un phénomène inévitable. Les taux de récidive après kératoplastie transfixiante sont rapportés à 17,8 % à 5 ans, 26 % à 8 ans et 56 % à 15 ans9). Comme l’opacité récurrente est généralement limitée à la couche superficielle, elle peut être éliminée par PTK, ce qui prolonge le délai avant une nouvelle greffe. Pour la LCD IIIA (type variant), un traitement n’est souvent pas nécessaire tant que la vision n’est pas significativement affectée.
QQuelle est l'efficacité de la PTK ?
A
La PTK peut éliminer efficacement les dépôts d’amyloïde superficiels, améliorant la vision et réduisant les érosions épithéliales récurrentes. Dans un cas de LCD IIIA, la meilleure acuité visuelle corrigée est passée de 20/400 à 20/50 après une PTK de 60 µm, sans récidive pendant 45 mois1). Chez les hétérozygotes, la récidive est lente, mais chez les homozygotes, une récidive précoce est observée. Les lésions profondes ne peuvent pas être éliminées par PTK, donc une DALK ou une kératoplastie transfixiante est nécessaire pour les opacités profondes7).
QLa LCD récidive-t-elle après une greffe de cornée ?
A
La récidive de la LCD après une greffe de cornée est inévitable. Les taux de récidive après kératoplastie transfixiante sont rapportés à 17,8 % à 5 ans, 26 % à 8 ans et 56 % à 15 ans9). Cependant, comme l’opacité récurrente est généralement limitée à la couche superficielle du greffon, elle peut être éliminée par PTK, prolongeant ainsi la durée de vie du greffon. La kératoplastie lamellaire profonde (DALK) présente un risque plus faible de rejet endothélial par rapport à la kératoplastie transfixiante et est considérée comme une nouvelle option de première intention7).
La pathologie centrale de la LCD1 est l’accumulation anormale de TGFBIp (kerato-épithéline, βig-h3). Le TGFBIp est normalement produit par l’épithélium cornéen, distribué dans toute l’épaisseur de la cornée, et dans le stroma, c’est une protéine structurale impliquée dans l’assemblage des fibres de collagène et l’adhésion cellulaire 5). La protéine anormale produite par la mutation R124C subit un mauvais repliement et une auto-agrégation, se déposant sous forme de fibrilles amyloïdes insolubles dans la couche de Bowman et le stroma superficiel. Au stade avancé, les dépôts s’étendent au stroma profond.
Les dépôts amyloïdes modifient les structures d’adhésion épithéliales de la cornée antérieure, provoquant une dégénérescence des cellules basales épithéliales et une dégénérescence de la couche épithéliale avec perte de la membrane de Bowman. Cette rupture structurelle constitue la base pathologique des érosions épithéliales cornéennes récurrentes.
Différenciation phénotypique selon le génotype TGFBI
Dans le gène TGFBI, les différences de site de mutation et d’acide aminé de substitution déterminent le tableau clinique. R124C provoque la LCD1, R124H la dystrophie cornéenne granuleuse de type II (type Avellino), et R124L la dystrophie cornéenne de Reis-Bücklers 5). Le mécanisme moléculaire par lequel une seule différence d’acide aminé détermine la substance déposée (amyloïde vs hyaline vs les deux) et le site de dépôt n’est pas complètement élucidé, mais on pense que le domaine de βig-h3 auquel appartient le site de mutation et son effet sur la stabilité du repliement sont essentiels.
Dans la LCD IIIA, des mutations profondes dominantes comme L527R produisent des lignes réticulées épaisses en forme de corde, et deviennent un type tardif sans trouble épithélial. La localisation stratifiée des dépôts peut s’expliquer par le gradient de sécrétion et de diffusion de βig-h3 des cellules productrices (cellules basales épithéliales) vers le stroma, et par les différences de stabilité de repliement de la protéine mutante. On pense que R124C privilégie la voie de formation de fibrilles amyloïdes à partir d’intermédiaires de repliement, accumulant l’amyloïde autour de la couche de Bowman5). En revanche, la mutation L527R forme une protéine mal repliée relativement stable, qui se dépose lentement dans le stroma plus profond.
Amyloïde cornéen postérieur et risque de chirurgie intraoculaire
Traditionnellement, on pensait que les dépôts amyloïdes de la LCD1 étaient limités à la cornée antérieure (couche de Bowman au stroma superficiel). Cependant, des études pathologiques récentes ont montré que des dépôts amyloïdes existent également dans la cornée postérieure près de la membrane de Descemet3). Les dépôts amyloïdes de la cornée postérieure peuvent affecter l’adhésion de la membrane de Descemet et contribuer à son décollement lors de la chirurgie de la cataracte3). On suggère que le même mécanisme par lequel les dépôts amyloïdes perturbent l’adhésion épithéliale dans la cornée antérieure agit également dans la partie postérieure 3).
Mécanisme moléculaire de l’amylose de type gelsoline
La gelsoline, molécule responsable de l’ancien LCD2 (syndrome de Meretoja), est présente à la fois dans le cytoplasme et l’espace extracellulaire. C’est une protéine qui participe à la motilité cellulaire, la division cellulaire et l’apoptose via la liaison à l’actine. La mutation classique D187N, appelée type finlandais, se manifeste par des dépôts cornéens en treillis et une neuropathie crânienne prédominante11). La nouvelle mutation p.Glu580Lys rapportée dans une famille slovène est située à la frontière des domaines G4-G5. Le remplacement de l’acide glutamique chargé négativement par la lysine chargée positivement provoque une répulsion électrostatique, réduisant la connectivité et la stabilité inter-domaines2). La gelsoline mutante subit un clivage anormal dans le plasma par la furine et la MT1-MMP, libérant des fragments précurseurs d’amyloïde de 8 kDa et 5 kDa. Ceux-ci se déposent dans la matrice cornéenne, la peau, les parois vasculaires, les nerfs périphériques et les glomérules rénaux, provoquant les symptômes multi-organiques caractéristiques du syndrome de Meretoja2,11). Les dépôts cornéens précèdent souvent les autres symptômes systémiques, ce qui peut permettre à l’ophtalmologiste de diagnostiquer la maladie en premier.
Des cas de LCD dus à des mutations de novo du gène TGFBI ont été rapportés1). Même en l’absence d’antécédents familiaux, la possibilité d’une mutation de novo doit être envisagée, et une confirmation par test génétique est recommandée1). La mutation L509P est rare mais peut se présenter sous des phénotypes variés, allant de la dystrophie cornéenne de Reis-Bücklers à la LCD IIIA1).
En plus des mutations classiques p.Asp187Asn/Tyr du gène GSN, une nouvelle mutation p.Glu580Lys a été rapportée, provoquant une amylose systémique avec dystrophie cornéenne en treillis, cutis laxa, arythmie cardiaque, néphropathie et neuropathie optique2).
Lésions cornéennes postérieures et gestion chirurgicale intraoculaire
Des dépôts amyloïdes ont été mis en évidence dans la cornée postérieure des patients atteints de LCD1, pouvant affecter l’adhésion de la membrane de Descemet3). Une attention particulière doit être portée au risque de décollement de la membrane de Descemet lors des chirurgies intraoculaires, notamment la cataracte.
Cette observation a des implications cliniques pour l’évaluation de l’indication de la chirurgie de la cataracte et la planification de la procédure chez les patients atteints de LCD1.
Des techniques chirurgicales plus précises, telles que la kératectomie lamellaire assistée par laser femtoseconde (femtosecond laser-assisted lamellar keratectomy, FLK) et la kératoplastie lamellaire assistée par laser femtoseconde (femtosecond laser-assisted lamellar keratoplasty, FALK), sont en cours de développement 12). Elles sont en train d’être positionnées comme des options complémentaires à la PTK conventionnelle, grâce à une meilleure régularité de la surface de résection et un contrôle de la profondeur hautement reproductible.
Étant donné que la mutation TGFBI est une mutation autosomique dominante de type gain de fonction, des siRNA spécifiques de l’allèle mutant, des oligonucléotides antisens et un knockout allèle-spécifique par CRISPR-Cas9 sont à l’étude au stade préclinique. La cornée est un organe cible favorable pour la thérapie génique car elle est accessible à l’administration locale et possède un privilège immunitaire. Cependant, aucune application clinique n’existe à ce jour, et toutes nécessitent une validation future de la sécurité et de l’efficacité à long terme.
Inhibiteurs de la formation d’amyloïde et chaperons moléculaires
Des petites molécules ciblant le processus d’agrégation de la TGFBIp et de la gelsoline mutante, des chaperons moléculaires (tels que les inducteurs de Hsp70) et des inhibiteurs de la liaison des fibrilles amyloïdes sont à l’étude au stade de la recherche fondamentale. Pour l’amylose systémique de type gelsoline, des médicaments inhibant l’étape de clivage de la gelsoline mutante dans le plasma sont évalués dans certains essais précliniques 2). À l’avenir, ces thérapies moléculaires ciblées sont attendues comme un traitement curatif remplaçant les résections physiques conventionnelles (PTK et greffe de cornée).
L’analyse protéomique de la cornée par spectrométrie de masse a montré que les dépôts de LCD1 contiennent non seulement de la TGFBIp mais aussi plusieurs autres protéines anormales qui pourraient co-précipiter. Des recherches sont en cours pour élucider la contribution pathologique de ces protéines co-précipitées en vue d’une future application clinique.
Ji YW, Ahn H, Shin KJ, Kim TI, Seo KY, Stulting RD, et al. De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. Journal of clinical medicine. 2022;11(11). doi:10.3390/jcm11113055. PMID:35683443; PMCID:PMC9181583.
Potrc M, Volk M, de Rosa M, Pizem J, Teran N, Jaklic H, Maver A, Drnovsek-Olup B, Bollati M, Vogelnik K, Hocevar A, Gornik A, Pfeifer V, Peterlin B, Hawlina M, Fakin A. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. Int J Mol Sci. 2021;22(3):1084.
Matsumoto A, Fukuoka H, Sotozono C. Descemet’s Membrane Detachment During Cataract Surgery in Lattice Corneal Dystrophy Type I: Histopathological Analysis of Posterior Corneal Involvement. Cureus. 2025;17(3):e81431. doi:10.7759/cureus.81431. PMID:40296953; PMCID:PMC12037203.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
Lisch W, Seitz B. The clinical landmarks of corneal dystrophies. Developments in ophthalmology. 2011;48:9-23. doi:10.1159/000324075. PMID:21540629.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern. San Francisco, CA: American Academy of Ophthalmology; 2024.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea. 2003;22(1):19-21.
Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969;1(4):314-324. PMID:4313418.
Kivelä T, Tarkkanen A, Frangione B, Ghiso J, Haltia M. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja’s syndrome. Invest Ophthalmol Vis Sci. 1994;35(10):3759-3769. PMID:8088963.
Steger B, Romano V, Biddolph S, Willoughby CE, Batterbury M, Kaye SB. Femtosecond laser-assisted lamellar keratectomy for corneal opacities secondary to anterior corneal dystrophies: an interventional case series. Cornea. 2016;35(1):6-13. PMID:26509759. doi:10.1097/ICO.0000000000000665.
Copiez le texte de l'article et collez-le dans l'assistant IA de votre choix.
Article copié dans le presse-papiers
Ouvrez un assistant IA ci-dessous et collez le texte copié dans la conversation.