전통적으로 “격자상 각막 이영양증 2형”이라고 불리던 유형은 전신성 겔솔린형 아밀로이드증(GSN-AMYL, Meretoja 증후군)의 안구 증상이며, 현재 IC3D 분류에서는 “가족성 아밀로이드증”으로 분류되어 고전적 LCD와 별도로 취급됩니다4,10). 1969년 핀란드의 Meretoja에 의해 기술된 이 증후군은 각막 격자상 혼탁 외에도 진행성 뇌신경 장애, 피부 이완, 전신 증상을 동반하는 유전성 질환입니다10,11). 임상 현장에서 두 질환의 감별이 중요하므로, 본 기사에서도 함께 기술합니다.
일본에서 빈도가 높은 TGFBI 관련 각막이영양증은 압도적으로 과립형 II형(Avellino형, R124H)이며, LCD1은 이에 비해 드뭅니다. 그러나 둘은 동일한 TGFBI 유전자의 단지 몇 염기 차이로 병형이 갈리므로, 임상 양상이 중복되는 경우 유전자 검사를 통한 확진이 바람직합니다. 일본에서 LCD 전체의 정확한 유병률은 보고되지 않았지만, 각막이영양증 전체 중에서는 비교적 드문 범주에 속합니다.
QLCD1과 Meretoja 증후군은 어떻게 다른가요?
A
LCD1은 TGFBI 유전자 돌연변이에 의한 각막에 국한된 아밀로이드 침착으로, 1020대에 동공 영역에서 발병하여 재발성 상피 미란을 자주 동반합니다. 반면 Meretoja 증후군(구 LCD2, GSN형)은 GSN(겔솔린) 유전자 돌연변이에 의한 전신성 아밀로이드증의 안구 증상으로, 3040대에 각막 주변부에서 발병하며 중심부의 투명성이 오래 유지됩니다. Meretoja 증후군에서는 피부 이완, 가면양 얼굴, 말초 신경병증, 심장 부정맥 등의 전신 증상을 동반합니다2,10). IC3D 제2판에서는 Meretoja 증후군이 격자상 각막이영양증과 독립적으로 분류됩니다4).
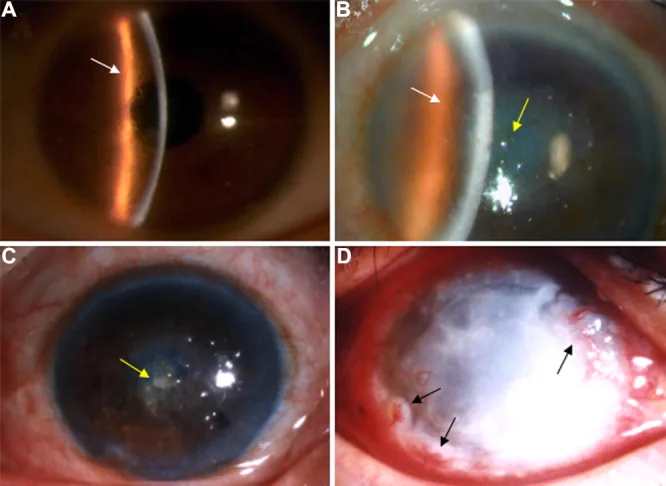
Liu Z, et al. An R124C mutation in TGFBI caused lattice corneal dystrophy type I with a variable phenotype in three Chinese families. Mol Vis. 2008. Figure 2. PMCID: PMC2443752. License: CC BY.
세극등 사진에서 각막 실질에 분지상의 격자선과 중심부 우세의 혼탁이 보입니다. 격자상 각막 이영양증의 대표적인 임상 소견을 보여주는 이미지입니다.
LCD1에서는 소아기에 대부분 무증상이며, 세극등 현미경의 투과 조명법으로 간신히 검출되는 미세 혼탁만 있습니다. 10~20대 이후에 재발성 각막 상피 미란(RCE)이 반복되어 기상 시 급성 안통, 눈부심, 눈물, 이물감이 반복됩니다. 30대경 각막 중앙부 실질 천층에 백색 혼탁이 현저해지고, 40대 이후 시력 저하가 진행됩니다.
LCD IIIA(변이형)에서는 상피 장애가 일반적으로 발생하지 않으며, 40세 이후 서서히 시력이 저하되는 것이 주된 증상입니다.
구 LCD2(Meretoja 증후군)에서는 안구 증상이 30~40대에 나타나지만, 심각한 시력 장애는 60대까지 지연되는 경우가 많습니다11). 눈꺼풀 피부 이완, 가면양 얼굴, 진행성 뇌신경 장애, 심장 부정맥 등 전신 증상이 선행하거나 동반되는 경우가 많습니다2,10).
격자상 선:실질 중층~심층에 굵고 긴 격자상 선, 때로는 수지상 분지를 나타냅니다. 직접 조명에서도 관찰 가능합니다.
표현형:①격자상 선만, ②소립상 침착만, ③둘의 혼합, 세 가지 패턴이 있습니다. 동일 개체 내에서 좌우안이 다른 표현형을 보이거나 편안성 증례도 있습니다.
상피:일반적으로 상피 장애는 발생하지 않습니다.
동형접합체:L527R 동형접합체에서는 격자상 선이 더 굵고 중앙의 과립상 침착이 크지만, R124H(과립상 II형)의 이형·동형 간 차이만큼 현저하지는 않습니다.
GSN형 (Meretoja)
격자상 선:섬세함이 부족한 소수의 격자상 침착이 주변부에서 방사상으로 나타납니다.
중앙 투명성:발병 후 장기간 중앙부의 투명성이 유지됩니다.
상피 미란:드뭅니다.
전신 소견:가면양 안면, 운동 장애를 동반한 돌출된 입술, 처진 귀, 안검 피부 이완증 등의 안면 변화를 나타냅니다2).
LCD1에서는 증례에 따라 중앙부의 원형 혼탁이 특히 심해지는 경우가 있으며, 56세 R124C 이형접합체에서 중앙 원형 혼탁으로 인해 각막 이식이 필요했던 보고가 있습니다.
Q소아에서도 LCD1을 진단할 수 있나요?
A
소아기 LCD1은 대부분 무증상이며, 직접 조명만으로는 이상을 찾기 어렵습니다. 세극등 현미경의 투과 조명법이나 반사 조명법을 이용한 상세한 관찰로 중앙 실질 천층의 미세한 점상~선상 혼탁을 확인할 수 있습니다. 재발성 각막 상피 미란을 반복하는 소아에서는 LCD1을 염두에 두고, 가족력 청취와 양친의 각막 검사를 포함한 평가가 권장됩니다. 확진에는 TGFBI 유전자 검사가 유용합니다.
고전적 돌연변이: D187N (핀란드형)이 가장 많고, p.Asp187Tyr도 보고됨10,11).
새로운 돌연변이: 슬로베니아 가족에서 보고된 p.Glu580Lys는 G4-G5 도메인 경계에 위치하며, 음전하에서 양전하로의 치환으로 정전기적 반발을 일으킴2).
임상 양상: 각막 격자 혼탁 외에도 피부 이완, 심장 부정맥, 신장 장애, 시신경 병증을 동반한 전신 아밀로이드증을 나타냄2).
유전 질환이므로 가족력이 가장 중요한 위험 인자입니다. 그러나 TGFBI에서는 de novo 돌연변이도 발생할 수 있으므로 가족력이 없다고 해서 배제할 수 없습니다1). 유전 양식은 상염색체 우성이며, 부모 중 한 명이 돌연변이 보유자이면 자녀에게 50% 확률로 전달됩니다. 성별 차이는 없으며, LCD1에서는 인종 차이가 명확하지 않지만 Meretoja 증후군은 핀란드에 다발 가계가 집적되는 것으로 알려져 있습니다11).
환경 요인의 기여는 명확하지 않으며, 본 질환의 발병 및 진행은 원칙적으로 유전자형에 의해 결정됩니다. 그러나 재발성 상피 미란의 빈도는 건조 환경, 콘택트렌즈 착용, 외상 등으로 악화될 수 있습니다. 굴절 교정 수술(LASIK, SMILE 등)은 TGFBI 관련 이영양증을 급속히 악화시킬 수 있으므로, 수술 전 선별 검사에서 가족력이 있는 증례에는 주의가 필요합니다5).
과립상 각막 이영양증 II형(Avellino형, TGFBI R124H): 일본에서 가장 많은 TGFBI 관련 이영양증이며, 과립상 침착과 격자상 선의 혼합상을 보입니다. LCD1과의 감별에는 유전자 검사가 확실합니다.
이차성 각막 아밀로이드증: 유전성이 아니며, 속눈썹난생, 원추각막 등 만성 안구표면 자극에 의해 이차적으로 아밀로이드가 침착됩니다. 가족력이 없고 기저 질환이 있는 것이 감별점입니다.
반점상 각막 이영양증: CHST6 유전자 이상에 의한 상염색체 열성 유전으로, 미만성 젖빛 유리 혼탁과 내피 이상을 동반합니다.
교양 적상 각막 이영양증: TACSTD2 유전자 이상에 의한 상염색체 열성 유전으로, 유백색의 교양 융기를 나타냅니다. 일본에서 비교적 흔합니다.
Q유전자 검사가 왜 중요한가요?
A
격자상 각막 이영양증은 표현형이 유사하더라도 원인 유전자와 돌연변이 부위가 다르면 진행 속도, 재발 빈도, 치료 선택, 전신 합병증 유무가 크게 달라집니다. TGFBI 돌연변이의 LCD1과 GSN 돌연변이의 Meretoja 증후군은 치료 방침과 전신 검사의 필요성이 근본적으로 다릅니다2,10). 또한 de novo 돌연변이가 있어 가족력만으로 병형을 결정할 수 없는 증례도 보고되어1), 유전자 검사가 확진과 병형 분류에 필수적입니다.
각막 표층에 아밀로이드 침착이 주된 LCD1에서 중심부 혼탁이 심하거나 재발성 각막 상피 미란이 반복되는 경우, 엑시머 레이저를 이용한 광치료 각막 절제술(PTK)이 일차 선택입니다7,8). 일반적으로 조기 재발은 없지만 시간이 지남에 따라 재발은 불가피하며, PTK 치료는 동일 안구에 대해 최대 2회까지 시행 가능합니다.
이형접합체에서는 재발이 느리고 재치료가 필요한 경우가 드물다. 동형접합체에서는 이형접합체에 비해 조기에 재발하는 경향이 있다. PTK 후 재발률은 다른 TGFBI 관련 이영양증과 마찬가지로 시간이 지남에 따라 증가하며, 장기 관찰에서 많은 증례에서 어떤 재발 소견이 확인된다8).
PTK의 유효성을 보여주는 증례로, TGFBI L509P의 de novo 돌연변이에 의한 LCD IIIA 증례에서 FD-OCT 유도 하에 60 µm의 PTK를 시행하여 최대교정시력(BCVA)이 20/400에서 20/50으로 개선되었다1). 수술 후 45개월 시점에서 시력 저하나 유의한 재발은 관찰되지 않았다1).
AAO의 각막 부종 및 혼탁 선호 진료 패턴에 따르면, 과립형 및 격자형 각막 이영양증에 대한 PTK는 ‘합리적 선택’이며 DALK나 전층 각막 이식으로의 전환을 지연시킬 수 있지만, 수술 후 헤이즈의 위험이 있다. 반복 시행 시 마이토마이신 C 병용이 재발성 흉터 및 실질 침착을 억제하는 수단으로 고려되며, 절제가 실질 전방 1/3을 초과하거나 잔여 베드가 250 µm 미만인 경우 각막확장증의 위험이 증가한다고 경고된다7).
재발을 반복하는 경우, 또는 혼탁이 실질 중층보다 깊은 층에 미치는 경우 각막 이식을 선택한다. LCD1에서는 보통 40세가 넘을 때까지 각막 이식의 적응이 되지 않는 경우가 많다. LCD에서는 각막 내피 세포는 원칙적으로 정상이므로, 혼탁의 깊이에 따라 수술 방식을 선택한다.
최근에는 거부 반응 위험 감소와 전층 각막 이식에 필적하는 시력 결과로 인해 DALK가 새로운 일차 선택으로 널리 사용되고 있습니다.
각막 이식 후 LCD 재발은 불가피하며, 전층 각막 이식 후 재발률은 5년 17.8%, 8년 26%, 15년 56%로 보고되었습니다 9). 재발 혼탁은 일반적으로 표층에 국한되므로 PTK로 제거할 수 있으며 재이식까지의 기간을 연장할 수 있습니다. LCD IIIA(변이형)의 경우 시력에 큰 영향이 없는 한 치료가 필요하지 않은 경우가 많습니다.
QPTK는 어느 정도 효과적입니까?
A
PTK는 표층의 아밀로이드 침착을 효과적으로 제거할 수 있으며 시력 개선과 재발성 상피 미란 감소를 얻을 수 있습니다. LCD IIIA 증례에서는 60 µm PTK 후 최대교정시력이 20/400에서 20/50으로 개선되었고 45개월 동안 재발이 없었다고 보고되었습니다 1). 이형접합체에서는 재발이 느리지만 동형접합체에서는 조기 재발이 나타납니다. 깊은 병변은 PTK로 제거할 수 없으므로 심층 혼탁에는 DALK 또는 전층 각막 이식이 필요합니다 7).
Q각막 이식 후 재발합니까?
A
각막 이식 후 LCD 재발은 피할 수 없습니다. 전층 각막 이식 후 재발률은 5년 17.8%, 8년 26%, 15년 56%로 보고되었습니다 9). 그러나 재발 혼탁은 일반적으로 이식편의 표층에 국한되므로 PTK로 제거할 수 있으며 이식편 수명을 연장할 수 있습니다. 심층판층각막이식술(DALK)은 전층 각막 이식에 비해 내피형 거부 반응 위험이 낮으며 새로운 일차 선택으로 주목받고 있습니다 7).
TGFBI 유전자에서 돌연변이 부위와 치환 아미노산의 차이가 임상 양상을 결정합니다. R124C는 LCD1을, R124H는 과립상 각막 이영양증 II형(Avellino형)을, R124L은 Reis-Bücklers 각막 이영양증을 유발합니다5). 단 하나의 아미노산 차이가 침착 물질(아밀로이드 vs 히알린 vs 둘 다)과 침착 부위를 결정하는 분자 기전은 완전히 밝혀지지 않았지만, 돌연변이 부위가 βig-h3의 어떤 도메인에 속하는지와 접힘 안정성에 미치는 영향이 핵심으로 여겨집니다.
LCD IIIA에서는 L527R 등의 심층 우세 돌연변이가 굵은 밧줄 모양의 격자선을 생성하고 상피 장애를 동반하지 않는 후발형이 됩니다. 침착물의 층별 국소화는 βig-h3의 생성 세포(상피 기저 세포)에서 실질 내로의 분비 및 확산 구배와 돌연변이 단백질의 접힘 안정성 차이로 설명될 수 있습니다. R124C는 접힘 중간체에서 아밀로이드 원섬유 형성으로 가는 경로를 우선하여 Bowman층 주변에 아밀로이드를 축적시키는 것으로 생각됩니다5). 반면 L527R 돌연변이는 비교적 안정한 잘못 접힌 단백질을 형성하여 더 깊은 실질층에 천천히 침착됩니다.
젤솔린(gelsolin)은 구형 LCD2(Meretoja 증후군)의 원인 분자로, 세포질과 세포외 모두에 존재하며 액틴 결합을 통해 세포 운동, 세포 분열, 세포 사멸에 관여하는 단백질입니다. 고전적인 D187N 돌연변이는 핀란드형이라고 불리며, 각막 격자 침착과 뇌신경 장애를 주된 표현형으로 나타냅니다11). 슬로베니아 가족에서 보고된 새로운 p.Glu580Lys 돌연변이는 G4-G5 도메인 경계에 위치하며, 음전하를 띤 글루탐산이 양전하를 띤 라이신으로 치환되어 정전기적 반발을 일으켜 도메인 간 연결성과 안정성을 감소시키는 것으로 생각됩니다2). 돌연변이 젤솔린은 혈장에서 퓨린(furin)과 MT1-MMP에 의해 비정상적으로 절단되어 8 kDa 및 5 kDa의 아밀로이드 전구체 단편을 방출합니다. 이들은 각막 기질, 피부, 혈관벽, 말초 신경, 신장 사구체에 침착되어 Meretoja 증후군에 특징적인 다기관 증상을 유발합니다2,11). 각막 침착은 종종 다른 전신 증상보다 먼저 나타나며, 안과 의사가 이 질환을 처음 진단하는 계기가 될 수 있습니다.
TGFBI 유전자의 de novo 돌연변이에 의한 LCD 발생이 보고되었습니다1). 가족력이 없는 경우에도 de novo 돌연변이 가능성을 고려하여 유전자 검사를 통한 확인이 권장됩니다1). L509P 돌연변이는 드물지만 Reis-Bücklers 각막 이영양증 유사에서 LCD IIIA 유사까지 다양한 표현형을 나타냅니다1).
GSN 유전자에서는 기존의 p.Asp187Asn/Tyr 돌연변이 외에 새로운 p.Glu580Lys 돌연변이가 보고되었으며, 각막 격자 이영양증, 피부 이완, 심장 부정맥, 신장 장애, 시신경 병증을 동반한 전신성 아밀로이드증을 유발하는 것으로 나타났습니다2).
TGFBI 돌연변이는 상염색체 우성의 기능획득 돌연변이이므로, 돌연변이 대립유전자 특이적 siRNA, 안티센스 올리고뉴클레오티드, CRISPR-Cas9에 의한 대립유전자 특이적 녹아웃이 전임상 연구 단계에서 검토되고 있습니다. 각막은 국소 투여가 가능하고 면역 특권을 가지므로 유전자 치료의 표적 기관으로 유리합니다. 그러나 현재 임상에 적용된 것은 없으며, 모두 향후 장기 안전성과 유효성 검증이 필요합니다.
TGFBIp 및 돌연변 겔솔린의 응집 과정을 표적으로 하는 저분자 화합물, 분자 샤페론(Hsp70 유도제 등), 아밀로이드 원섬유 결합 억제제가 기초 연구 단계에서 검토되고 있습니다. 전신성 겔솔린형 아밀로이드증에 대해서는 혈장 내 돌연변이 겔솔린 절단 단계를 억제하는 약물이 일부 전임상 시험에서 평가되고 있습니다2). 향후 이러한 분자 표적 치료가 기존의 물리적 절제(PTK 및 각막 이식)를 대체하는 근본적인 치료법으로 기대됩니다.
Ji YW, Ahn H, Shin KJ, Kim TI, Seo KY, Stulting RD, et al. De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. Journal of clinical medicine. 2022;11(11). doi:10.3390/jcm11113055. PMID:35683443; PMCID:PMC9181583.
Potrc M, Volk M, de Rosa M, Pizem J, Teran N, Jaklic H, Maver A, Drnovsek-Olup B, Bollati M, Vogelnik K, Hocevar A, Gornik A, Pfeifer V, Peterlin B, Hawlina M, Fakin A. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. Int J Mol Sci. 2021;22(3):1084.
Matsumoto A, Fukuoka H, Sotozono C. Descemet’s Membrane Detachment During Cataract Surgery in Lattice Corneal Dystrophy Type I: Histopathological Analysis of Posterior Corneal Involvement. Cureus. 2025;17(3):e81431. doi:10.7759/cureus.81431. PMID:40296953; PMCID:PMC12037203.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
Lisch W, Seitz B. The clinical landmarks of corneal dystrophies. Developments in ophthalmology. 2011;48:9-23. doi:10.1159/000324075. PMID:21540629.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern. San Francisco, CA: American Academy of Ophthalmology; 2024.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea. 2003;22(1):19-21.
Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969;1(4):314-324. PMID:4313418.
Kivelä T, Tarkkanen A, Frangione B, Ghiso J, Haltia M. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja’s syndrome. Invest Ophthalmol Vis Sci. 1994;35(10):3759-3769. PMID:8088963.
Steger B, Romano V, Biddolph S, Willoughby CE, Batterbury M, Kaye SB. Femtosecond laser-assisted lamellar keratectomy for corneal opacities secondary to anterior corneal dystrophies: an interventional case series. Cornea. 2016;35(1):6-13. PMID:26509759. doi:10.1097/ICO.0000000000000665.