Lattice kornea distrofisi (LCD), kornea stromasında amiloid birikimi ile kafes benzeri çizgisel opasiteler oluşturan kalıtsal bir kornea distrofisidir. 1890’larda tanımlanmış eski bir hastalıktır ve IC3D 2. baskı klinik-genetik sınıflamasında LCD1 ve varyantları (eski tip 3, 3A, 1/3A, 4) olarak birleştirilmiştir4).
LCD1, granüler kornea distrofisi, Reis-Bücklers kornea distrofisi ve Thiel-Behnke kornea distrofisi, “TGFBI ile ilişkili distrofiler” olarak bir hastalık grubu oluşturur. Sorumlu gen TGFBI (transforming growth factor beta-induced gene) 5. kromozomun uzun kolunda (5q31) yer alır ve otozomal dominant kalıtım gösterir. TGFBI proteini (TGFBIp, kerato-epitelin, βig-h3) kornea epiteli tarafından üretilir ve korneanın tüm katmanlarında bulunur. Kornea stromasında kollajen liflerinin yapısında rol oynar. Aynı genin mutasyonları, mutasyon bölgesi ve amino asit değişimine bağlı olarak biriken madde (hialin veya amiloid) ve klinik görünüm açısından büyük farklılık gösterir5).
LCD1’in temel mutasyonu, TGFBI genindeki 124. argininin sistein ile yer değiştirdiği R124C’dir. Varyant tip LCD IIIA’da L527R gibi mutasyonlar rapor edilmiştir.
Korneada biriken anormal protein, Kongo kırmızısı boyaması ile kırmızı renk alır ve polarize mikroskop altında karakteristik elma yeşili çift kırılım göstererek amiloid olduğu doğrulanır. Bu bulgu, 19. yüzyıldan beri klasik amiloidoz doku tanı göstergesidir6).
Eskiden “latis kornea distrofisi tip 2” olarak adlandırılan tip, sistemik gelsolin tipi amiloidozun (GSN-AMYL, Meretoja sendromu) bir göz bulgusudur ve mevcut IC3D sınıflamasında “familyal amiloidoz” olarak sınıflandırılır, klasik LCD’den bağımsız olarak ele alınır4,10). 1969’da Finlandiyalı Meretoja tarafından tanımlanan bu sendrom, korneada latis opasitelerine ek olarak ilerleyici kraniyal nöropati, cilt gevşekliği ve sistemik semptomlarla seyreden kalıtsal bir hastalıktır10,11). Klinik pratikte ayırıcı tanı önemli olduğundan, bu makalede her iki durum da birlikte sunulmaktadır.
Japonya’da sık görülen TGFBI ile ilişkili distrofiler arasında granüler tip II (Avellino tipi, R124H) ezici çoğunluğu oluştururken, LCD1 buna kıyasla daha az yaygındır. Ancak her ikisi de aynı TGFBI genindeki yalnızca birkaç baz çifti farkıyla ayrıldığından, klinik tabloları örtüşen vakalarda genetik test ile doğrulama önerilir. Japonya’da LCD’nin genel prevalansı rapor edilmemiş olmakla birlikte, tüm kornea distrofileri arasında nispeten nadir bir gruptur.
QLCD1 ve Meretoja sendromu arasındaki fark nedir?
A
LCD1, TGFBI gen mutasyonuna bağlı korneaya sınırlı amiloid birikimidir; 10-20 yaşlarında pupiller alandan başlar ve sıklıkla tekrarlayan epitel erozyonları eşlik eder. Öte yandan Meretoja sendromu (eski adıyla LCD2, GSN tipi), GSN (gelsolin) gen mutasyonuna bağlı sistemik amiloidozun göz bulgusudur; 30-40 yaşlarında periferik korneadan başlar ve merkezi saydamlık uzun süre korunur. Meretoja sendromunda cilt gevşekliği, maske yüz görünümü, periferik nöropati ve kardiyak aritmi gibi sistemik bulgular eşlik eder2,10). IC3D 2. baskısında Meretoja sendromu, kafes kornea distrofisinden bağımsız olarak sınıflandırılmıştır4).
Liu Z, et al. An R124C mutation in TGFBI caused lattice corneal dystrophy type I with a variable phenotype in three Chinese families. Mol Vis. 2008. Figure 2. PMCID: PMC2443752. License: CC BY.
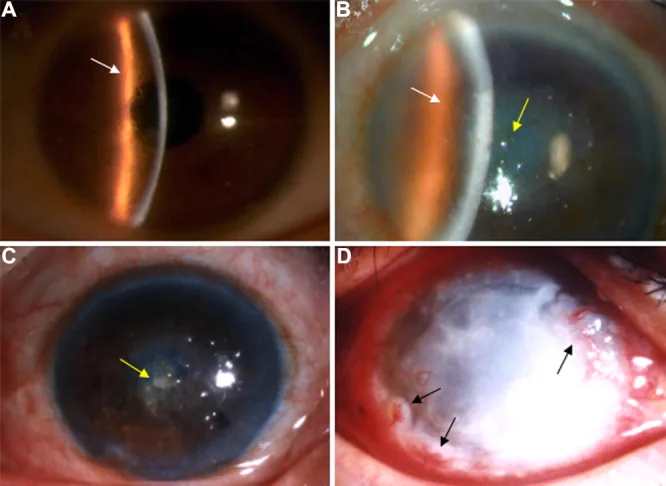
Yarık lamba fotoğrafında, kornea stromasında dallanan çizgisel çizgiler ve merkezde baskın bulanıklık görülüyor. Lattice kornea distrofisinin tipik klinik bulgusunu gösteren bir görüntüdür.
LCD1’de çocukluk döneminde çoğu asemptomatiktir, sadece yarık lamba mikroskobunun retroillüminasyon yöntemiyle zorlukla tespit edilen ince bulanıklıklar vardır. 10-20’li yaşlardan itibaren tekrarlayan kornea epitel erozyonları (RCE) görülür ve uyanma sırasında ani göz ağrısı, fotofobi, sulanma ve yabancı cisim hissi tekrarlar. 30’lu yaşlarda kornea merkezindeki stromanın yüzeysel tabakasında beyaz bulanıklık belirginleşir ve 40’lı yaşlardan sonra görme azalması ilerler.
LCD IIIA (varyant tip) genellikle epitel hasarına neden olmaz ve 40 yaşından sonra yavaş ilerleyen görme azalması ana şikayettir.
Eski LCD2 (Meretoja sendromu) göz belirtileri 30-40’lı yaşlarda ortaya çıkar, ancak ciddi görme bozukluğu genellikle 60’lı yaşlara kadar gecikir 11). Göz kapağı derisinde gevşeklik, maske yüz görünümü, ilerleyici kraniyal nöropati, kardiyak aritmi gibi sistemik belirtiler sıklıkla önceden veya eş zamanlı olarak ortaya çıkar 2,10).
Hastalık tipine göre yarık lamba mikroskobu bulguları aşağıda gösterilmiştir.
LCD1 (Klasik Tip)
Başlangıç yeri: Her iki gözün pupil alanında Bowman tabakası ile stromanın yüzeysel tabakasında ince noktasal ve çizgisel bulanıklıklar olarak ortaya çıkar.
Kafes şeklinde çizgiler: Çift konturlu ipliksi-çizgisel bulanıklıklar birbirine dolanarak ağsı ve yıldız şeklinde bulanıklıklar oluşturur.
İlerlemiş evre: Merkezi korneada yumurta sarısı veya yuvarlak şekilli süt beyazı bulanıklık oluşur.
Retroillüminasyon: Doğrudan aydınlatmada zor görülen yarı saydam ince kafes şeklindeki çizgiler net bir şekilde ortaya çıkar.
Floresein boyama: Epitel yapışmasının azalması nedeniyle yüzey pürüzlü hale gelir.
Tekrarlayan epitel erozyonları: Birikintiler epitel bazal hücrelerine ve Bowman tabakasına kadar uzandığı için sık görülür.
LCD IIIA (Varyant Tip)
Çizgisel çizgiler: Stromanın orta-derin katmanlarında kalın, uzun çizgisel çizgiler, bazen dendritik dallanma gösterir. Direkt aydınlatma ile de gözlemlenebilir.
Fenotipler: ① sadece çizgisel çizgiler, ② sadece küçük granüler birikintiler, ③ her ikisinin karışımı olmak üzere 3 patern vardır. Aynı bireyde sağ ve sol gözler farklı fenotipler gösterebilir veya tek taraflı olgular da mevcuttur.
Epitel: Genellikle epitel hasarı oluşmaz.
Homozigotlar: L527R homozigotlarında çizgisel çizgiler daha kalın ve merkezi granüler birikintiler daha büyüktür, ancak R124H (granüler tip II) heterozigot-homozigot arasındaki fark kadar belirgin değildir.
GSN tipi (Meretoja)
Çizgisel çizgiler: İncelikten yoksun, az sayıda çizgisel birikinti periferden radyal olarak ortaya çıkar.
Merkezi şeffaflık: Başlangıçtan sonra uzun süre merkezi şeffaflık korunur.
Epitel erozyonu: Nadirdir.
Sistemik bulgular: Maske yüz görünümü, hareket bozukluğu ile birlikte belirgin dudaklar, sarkık kulaklar, göz kapağı gevşekliği gibi yüz değişiklikleri görülür2).
LCD1’de bazı olgularda merkezi yuvarlak bulanıklık özellikle belirgin olabilir; 56 yaşında R124C heterozigot bir olguda merkezi yuvarlak bulanıklık nedeniyle kornea nakli endikasyonu konulduğu bildirilmiştir.
QÇocuklarda LCD1 tanısı konulabilir mi?
A
Çocukluk çağındaki LCD1 genellikle asemptomatiktir ve sadece direkt aydınlatma ile anormallik bulmak zordur. Yarık lamba mikroskobunun retroillüminasyon veya transillüminasyon yöntemleriyle detaylı inceleme, merkezi stroma yüzeyel katmanlarında ince noktasal-çizgisel bulanıklıkları ortaya çıkarabilir. Tekrarlayan kornea epitel erozyonu olan çocuklarda LCD1 akılda tutulmalı, aile öyküsü alınması ve ebeveynlerin kornea muayenesini içeren değerlendirme önerilir. Kesin tanı için TGFBI gen testi faydalıdır.
Latis kornea distrofisinin sorumlu genleri ve tipik mutasyonları aşağıda özetlenmiştir.
TGFBI ilişkili (LCD1, LCD IIIA, LCD IV)
Gen lokusu: 5q31 (TGFBI geni).
Kalıtım şekli: Otozomal dominant kalıtımdır.
LCD1 tipik mutasyon: R124C (Arg124Cys) en sık görülenidir5).
LCD IIIA tipik mutasyon: L527R (Leu527Arg) gibi mutasyonlar bildirilmiştir. Homozigot örnekler de mevcuttur.
De novo mutasyon: TGFBI L509P de novo mutasyonunun LCD IIIA fenotipi gösterdiği bir olgu bildirilmiştir1). Ebeveynlerde mutasyon yokken, çocuklardan birinde mutasyon kalıtılmıştır1).
TGFBIp’nin rolü: Kornea epiteli tarafından üretilir ve korneanın tüm katmanlarına dağılır; stromada kollajen lif yapısına katkıda bulunur5).
GSN ilişkili (Meretoja sendromu, eski LCD2)
Gen lokusu: 9q34 (GSN geni, gelsolin).
Kalıtım şekli: Otozomal dominant kalıtımdır.
Klasik mutasyon: D187N (Fin tipi) en yaygın olup, p.Asp187Tyr de bildirilmiştir10,11).
Yeni mutasyon: Sloven bir ailede bildirilen p.Glu580Lys, G4-G5 alan sınırında yer alır ve negatif yükten pozitif yüke değişimle elektrostatik itmeye neden olur2).
Klinik görünüm: Korneada kafes benzeri bulanıklığa ek olarak, cilt gevşekliği, kalp aritmisi, böbrek hasarı ve optik nöropati ile seyreden sistemik amiloidoz görülür2).
Kalıtsal bir hastalık olduğu için aile öyküsü en önemli risk faktörüdür. Ancak TGFBI’da de novo mutasyonlar oluşabileceğinden, aile öyküsü olmaması hastalığı dışlamaz1). Kalıtım şekli otozomal dominanttır; ebeveynlerden biri mutasyon taşıyıcısıysa çocuğa %50 olasılıkla geçer. Cinsiyet farkı yoktur ve LCD1’de ırk farkı belirgin değildir, ancak Meretoja sendromu Finlandiya’da yoğun aile kümelenmesiyle bilinir11).
Çevresel faktörlerin katkısı net değildir; hastalığın başlangıcı ve ilerlemesi prensipte genotip tarafından belirlenir. Ancak tekrarlayan epitel erozyonlarının sıklığı kuru ortam, kontakt lens kullanımı veya travma ile artabilir. Refraktif cerrahi (LASIK, SMILE vb.) TGFBI ile ilişkili distrofilerin hızlı kötüleşmesine neden olabilir; bu nedenle aile öyküsü olan vakalarda ameliyat öncesi taramada dikkatli olunmalıdır5).
LCD1, varyant tipler ve GSN tipinin ayırıcı tanısı için yarık lamba bulguları, doku bulguları ve genetik bulgular bir arada değerlendirilmelidir.
Klinik Testler
Yarık lamba mikroskobu: Doğrudan aydınlatmada erken dönemdeki kafes benzeri çizgiler gözden kaçabilir. Transillüminasyon yöntemiyle pupilla arka planında ince bulanıklıklar, retroillüminasyon yöntemiyle yarı saydam ince kafes çizgileri tespit edilir.
Floresein boyama: LCD1’de epitel adezyonunun azalmasına bağlı olarak boyanın yayılımı pürüzlü olur. Epitel erozyonunun yaygınlığını değerlendirmede de faydalıdır.
Ön segment optik koherens tomografi (Ön segment OCT): Birikintilerin katman bazında derinliğini kantitatif olarak değerlendirebilir. FD-OCT ile lezyon derinliğinin ölçümü, PTK’nın eksizyon derinliğinin belirlenmesinde faydalıdır1).
Kornea konfokal mikroskobu: Stromadaki birikintiler hücresel düzeyde gözlemlenebilir.
Kesin Tanı
Genetik test: TGFBI geni ve GSN genindeki mutasyonların saptanması ile hastalık tipi belirlenir. Aynı fenotipe sahip olsa bile farklı mutasyonlar nüks ve ilerleme hızını değiştirdiğinden, tedavi planlaması için de doğrudan önemlidir.
Patolojik inceleme: Kongo kırmızısı boyaması ile kırmızı renk alması ve polarize mikroskop altında elma yeşili çift kırılım göstermesi ile amiloid olduğu doğrulanır6).
İmmünohistokimya: Anti-TGFBIp antikoru ve anti-gelsolin antikoru ile hastalık tiplerinin ayırıcı tanısı mümkündür.
Aile öyküsü alınması: Otozomal dominant kalıtım nedeniyle ebeveynler ve kardeşlerdeki kornea bulgularının incelenmesi tanıyı destekler.
Granüler kornea distrofisi tip II (Avellino tipi, TGFBI R124H): Japonya’da en sık görülen TGFBI ilişkili distrofidir ve granüler birikintiler ile kafes çizgilerinin karışık görünümünü gösterir. LCD1 ile ayırıcı tanıda genetik test kesin sonuç verir.
Sekonder korneal amiloidoz: Kalıtsal olmayıp, trikiyazis veya keratokonus gibi kronik oküler yüzey irritasyonu zemininde sekonder olarak amiloid birikir. Aile öyküsü olmaması ve altta yatan bir hastalığın bulunması ayırt edici noktalardır.
Maküler kornea distrofisi: CHST6 gen mutasyonuna bağlı otozomal resesif geçişli olup, diffüz buzlu cam opasitesi ve endotel anormallikleri eşlik eder.
Jelatinöz damla benzeri kornea distrofisi: TACSTD2 gen mutasyonuna bağlı otozomal resesif geçişli olup, süt beyazı jelatinimsi kabarıklıklar şeklinde görülür. Japonya’da nispeten sık görülür.
QGenetik test neden önemlidir?
A
Lattice kornea distrofisinde fenotip benzer olsa bile, sorumlu gen ve mutasyon bölgesi farklı olduğunda ilerleme hızı, nüks sıklığı, tedavi seçimi ve sistemik komplikasyonların varlığı büyük ölçüde değişir. TGFBI mutasyonlu LCD1 ile GSN mutasyonlu Meretoja sendromunda tedavi yaklaşımı ve sistemik tarama gerekliliği temelde farklıdır2,10). Ayrıca, aile öyküsü olmaksızın de novo mutasyonlar bildirilmiştir1) ve genetik test, kesin tanı ve alt tip sınıflandırması için vazgeçilmezdir.
Çocukluk-genç erişkinlik döneminde asemptomatik veya sadece ince opasitelerin olduğu evrede takip önerilir. Altı ayda bir veya yılda bir yapılan yarık lamba muayenesi ile ilerleme değerlendirilir.
LCD1’in temel semptomu olan tekrarlayan epitel erozyonları için ilk basamak aşağıdaki konservatif tedavidir.
Atak tedavisi: Terapötik yumuşak kontakt lensin sürekli kullanımı ile kornea epiteli korunur. İkincil enfeksiyonu önlemek için antibakteriyel damlalar eklenir. Kayganlaştırma ve epitel koruması için göz merhemi uygulanır.
Nüks önleme: Yatmadan önce göz merhemi uygulanması RCE ataklarının tekrarlamasını baskılamada etkilidir. Kuru ortamlarda gündüzleri de suni gözyaşı veya kayganlaştırıcılar kullanılır.
Kornea yüzeyel amiloid birikiminin baskın olduğu LCD1’de, santral opasite belirginse veya tekrarlayan epitel erozyonları sık görülüyorsa, excimer lazer ile fototerapötik keratektomi (PTK) ilk seçenektir7,8). Genellikle erken nüks görülmez, ancak zamanla nüks kaçınılmazdır ve PTK aynı göze en fazla iki kez uygulanabilir.
Heterozigotlarda nüks yavaştır ve yeniden tedavi gerektiren vakalar azdır. Homozigotlarda ise heterozigotlara kıyasla daha erken nüksetme eğilimi vardır. PTK sonrası nüks oranı, diğer TGFBI ile ilişkili distrofilerde olduğu gibi zamanla artar ve uzun süreli takipte birçok vakada bir miktar nüks bulgusu tespit edilir8).
PTK’nın etkinliğini gösteren bir vaka olarak, TGFBI L509P de novo mutasyonuna bağlı LCD IIIA’lı bir hastada FD-OCT kılavuzluğunda 60 µm PTK uygulanmış ve en iyi düzeltilmiş görme keskinliği (BCVA) 20/400’den 20/50’ye iyileşmiştir1). Ameliyat sonrası 45. ayda görme azalması veya anlamlı nüks gözlenmemiştir1).
AAO’nun Kornea Ödemi ve Opasitesi Preferred Practice Pattern’ine göre, granüler ve latis kornea distrofilerinde PTK ‘makul bir seçenek’tir ve DALK veya penetran keratoplastiye geçişi geciktirebilir, ancak postoperatif hale riski vardır. Tekrarlanan uygulamalarda mitomisin C kullanımı, tekrarlayan skar veya stromal birikintileri baskılamak için bir yöntem olarak değerlendirilmektedir; ablasyon stromanın ön üçte birini aştığında veya rezidüel yatak 250 µm’den az olduğunda kornea ektazisi riskinin arttığı uyarısı yapılmaktadır7).
Tekrarlayan nüksler veya opasitenin stromanın orta-derin katmanlarına uzandığı durumlarda kornea nakli tercih edilir. LCD1’de genellikle 40 yaşına kadar kornea nakli endikasyonu konmaz. LCD’de kornea endotel hücreleri prensip olarak normal olduğundan, opasitenin derinliğine göre cerrahi yöntem seçilir.
Yüksek görme düzelme potansiyeli, ancak red ve nüks riski
Son yıllarda, red reaksiyonu riskini azaltması ve tam kat kornea nakline eşdeğer görme sonuçları sağlaması nedeniyle DALK, yeni birinci seçenek olarak yaygın şekilde kullanılmaktadır.
Kornea nakli sonrası LCD nüksü kaçınılmaz bir olgudur ve tam kat kornea nakli sonrası nüks oranları 5 yılda %17,8, 8 yılda %26, 15 yılda %56 olarak bildirilmiştir9). Nüks bulanıklığı genellikle yüzeyel tabakayla sınırlı olduğundan PTK ile temizlenebilir ve yeniden nakil süresi uzatılabilir. LCD IIIA (varyant tip) için ise görme üzerinde belirgin bir etki oluşmadıkça tedavi gerekmeyebilir.
QPTK ne kadar etkilidir?
A
PTK, yüzeyel amiloid birikimini etkili bir şekilde temizleyebilir ve görme iyileşmesi ile tekrarlayan epitel erozyonlarının azalmasını sağlar. LCD IIIA vakalarında 60 µm PTK sonrası en iyi düzeltilmiş görme keskinliğinin 20/400’den 20/50’ye iyileştiği ve 45 ay boyunca nüks olmadığı bildirilmiştir1). Heterozigotlarda nüks yavaş seyrederken, homozigotlarda erken nüks görülür. Derin lezyonlar PTK ile temizlenemediğinden, derin bulanıklık için DALK veya tam kat kornea nakli gerekir7).
QKornea naklinden sonra nüks olur mu?
A
Kornea nakli sonrası LCD nüksü kaçınılmazdır. Tam kat kornea nakli sonrası nüks oranları 5 yılda %17,8, 8 yılda %26, 15 yılda %56 olarak bildirilmiştir9). Ancak nüks bulanıklığı genellikle greftin yüzeyel tabakasıyla sınırlı olduğundan PTK ile temizlenebilir ve greft ömrü uzatılabilir. Derin lameller kornea nakli (DALK), tam kat kornea nakline kıyasla endotelyal red reaksiyonu riskini azaltır ve yeni birinci seçenek olarak dikkat çekmektedir7).
LCD1’in patogenezinin merkezinde TGFBIp’nin (kerato-epitelin, βig-h3) anormal birikimi yer alır. TGFBIp normalde kornea epiteli tarafından üretilir, korneanın tüm katmanlarına dağılır ve stromada kollajen lif yapısı ile hücre yapışmasında rol oynayan bir yapısal proteindir 5). R124C mutasyonu ile üretilen anormal protein, yanlış katlanma ve kendi kendine agregasyona uğrayarak çözünmeyen amiloid fibrilleri halinde Bowman tabakası ve stromanın yüzeysel kısmında birikir. İlerlemiş evrede birikim stromanın derin katmanlarına yayılır.
Amiloid birikimi, ön korneada epitel yapışma yapılarında değişikliğe neden olur ve epitel bazal hücrelerinin dejenerasyonu, Bowman membranının kaybı ile birlikte epitel tabakasında dejenerasyona yol açar. Bu yapısal bozulma, tekrarlayan kornea epitel erozyonlarının patofizyolojik temelini oluşturur.
TGFBI geninde mutasyon bölgesi ve değiştirilen amino asit farkı klinik tabloyu belirler. R124C LCD1’e, R124H granüler kornea distrofisi tip II’ye (Avellino tipi), R124L ise Reis-Bücklers kornea distrofisine neden olur 5). Sadece bir amino asit farkının birikim maddesini (amiloid vs hiyalin vs her ikisi) ve birikim bölgesini belirlemesinin moleküler mekanizması tam olarak aydınlatılamamıştır, ancak mutasyon bölgesinin βig-h3’ün hangi domainine ait olduğu ve katlanma stabilitesine etkisinin anahtar rol oynadığı düşünülmektedir.
LCD IIIA’da L527R gibi derin tabaka baskın mutasyonlar kalın ip benzeri kafes çizgileri oluşturur ve epitel hasarı olmayan geç başlangıçlı bir tip ortaya çıkar. Birikintilerin katmanlara göre lokalizasyonu, βig-h3’ü üreten hücrelerden (epitel bazal hücreleri) stromaya salgılanma ve difüzyon gradyanı ile mutant proteinin katlanma stabilitesindeki farklılıklarla açıklanabilir. R124C’nin katlanma ara ürünlerinden amiloid fibril oluşumuna giden yolu tercih ettiği ve Bowman tabakası çevresinde amiloid biriktirdiği düşünülmektedir 5). Öte yandan, L527R mutasyonu nispeten stabil bir yanlış katlanmış protein oluşturur ve daha derin stromada yavaşça birikir.
Geleneksel olarak LCD1’de amiloid birikiminin ön kornea (Bowman tabakası ve yüzeysel stroma) ile sınırlı olduğu düşünülüyordu. Ancak son yıllardaki patolojik incelemeler, Descemet membranı yakınındaki arka korneada da amiloid birikimi olduğunu göstermiştir 3). Arka korneadaki amiloid birikiminin Descemet membranının yapışmasını etkileyerek katarakt cerrahisi sırasında Descemet membranı dekolmanına katkıda bulunabileceği belirtilmiştir 3). Ön korneada amiloid birikiminin epitel yapışmasını bozmasına benzer bir mekanizmanın arka kısımda da etkili olduğu ileri sürülmektedir 3).
Eski LCD2 (Meretoja sendromu) etken molekülü gelsolin, sitoplazma ve hücre dışında bulunan, aktin bağlanması yoluyla hücre hareketi, hücre bölünmesi ve apoptozda rol oynayan bir proteindir. Klasik D187N mutasyonu, Finlandiya tipi olarak adlandırılan, korneal kafes benzeri birikimler ve kraniyal nöropati ile karakterize bir fenotip sergiler 11). Slovenya ailesinde bildirilen yeni p.Glu580Lys mutasyonu, G4-G5 alan sınırında yer alır ve negatif yüklü glutamattan pozitif yüklü lizine değişim, elektrostatik itmeye neden olarak alanlar arası bağlantı ve stabiliteyi azaltır 2). Mutant gelsolin, plazmada furin ve MT1-MMP tarafından anormal kesime uğrar ve 8 kDa ve 5 kDa’lık amiloid öncü fragmanları salınır. Bunlar kornea stroması, deri, damar duvarı, periferik sinir ve böbrek glomerüllerinde birikerek Meretoja sendromunun karakteristik çoklu organ semptomlarına yol açar 2,11). Korneal birikimler genellikle diğer sistemik semptomlardan önce gelir ve göz doktorunun bu hastalığı ilk teşhis etmesine olanak sağlayabilir.
TGFBI genindeki de novo mutasyonlara bağlı LCD gelişimi bildirilmiştir 1). Aile öyküsü olmayan vakalarda bile de novo mutasyon olasılığı düşünülmeli ve genetik test ile doğrulama önerilir 1). L509P mutasyonu nadirdir ancak Reis-Bücklers kornea distrofisi benzerinden LCD IIIA benzerine kadar değişen fenotipler sergiler 1).
GSN geninde, geleneksel p.Asp187Asn/Tyr mutasyonlarına ek olarak, yeni bir p.Glu580Lys mutasyonu bildirilmiş ve korneal kafes distrofisi, deri gevşekliği, kardiyak aritmi, böbrek yetmezliği ve optik nöropati ile seyreden sistemik amiloidoz ile ilişkilendirilmiştir 2).
Arka Kornea Lezyonları ve İç Göz Cerrahisi Yönetimi
LCD1 hastalarının arka korneasında amiloid birikimleri bulunduğu ve bunun Descemet membran yapışmasını etkileyebileceği patolojik olarak gösterilmiştir 3). Katarakt cerrahisi dahil iç göz cerrahisi sırasında Descemet membran dekolmanı riskine dikkat edilmelidir.
Bu bulgu, LCD1 hastalarında katarakt cerrahisi endikasyon değerlendirmesi ve cerrahi planlama için klinik öneme sahiptir.
Femtosaniye lazer destekli lameller keratektomi (FLK) ve femtosaniye lazer destekli lameller keratoplasti (FALK) gibi daha hassas cerrahi tekniklerin geliştirilmesi devam etmektedir 12). Bunlar, kesi yüzeyinin düzgünlüğünü artırması ve yüksek tekrarlanabilirlikte derinlik kontrolü sağlaması nedeniyle geleneksel PTK’yı tamamlayıcı seçenekler olarak konumlandırılmaktadır.
TGFBI mutasyonu otozomal dominant fonksiyon kazancı mutasyonu olduğundan, mutant alele özgü siRNA, antisense oligonükleotitler ve CRISPR-Cas9 ile alele özgü nakavt, preklinik çalışma aşamasında değerlendirilmektedir. Kornea, lokal uygulamaya uygun olması ve bağışıklık ayrıcalığına sahip olması nedeniyle gen tedavisi için avantajlı bir hedef organdır. Ancak şu anda klinik uygulamada olan bir tedavi yoktur ve tümü için gelecekte uzun dönem güvenlik ve etkinlik doğrulaması gereklidir.
Amiloid Oluşum İnhibitörleri ve Moleküler Şaperonlar
TGFBIp ve mutant gelsolinin agregasyon sürecini hedef alan düşük molekül ağırlıklı bileşikler, moleküler şaperonlar (Hsp70 indükleyicileri gibi) ve amiloid fibril bağlanma inhibitörleri temel araştırma aşamasında değerlendirilmektedir. Sistemik gelsolin tipi amiloidoz için, plazmadaki mutant gelsolinin kesilme aşamasını inhibe eden ilaçlar bazı preklinik çalışmalarda değerlendirilmiştir 2). Gelecekte bu tür moleküler hedefli tedavilerin, geleneksel fiziksel eksizyona (PTK, kornea nakli) alternatif olarak köklü bir tedavi olması beklenmektedir.
Kütle spektrometrisi kullanılarak yapılan kornea proteom analizi, LCD1 birikintilerinde yalnızca TGFBIp değil, aynı zamanda birden fazla anormal proteinin birlikte çökeldiğini göstermiştir. Gelecekteki klinik uygulamaya yönelik olarak, bu birlikte çökelen proteinlerin hastalık patogenezine katkısının aydınlatılması çalışmaları devam etmektedir.
Ji YW, Ahn H, Shin KJ, Kim TI, Seo KY, Stulting RD, et al. De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. Journal of clinical medicine. 2022;11(11). doi:10.3390/jcm11113055. PMID:35683443; PMCID:PMC9181583.
Potrc M, Volk M, de Rosa M, Pizem J, Teran N, Jaklic H, Maver A, Drnovsek-Olup B, Bollati M, Vogelnik K, Hocevar A, Gornik A, Pfeifer V, Peterlin B, Hawlina M, Fakin A. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. Int J Mol Sci. 2021;22(3):1084.
Matsumoto A, Fukuoka H, Sotozono C. Descemet’s Membrane Detachment During Cataract Surgery in Lattice Corneal Dystrophy Type I: Histopathological Analysis of Posterior Corneal Involvement. Cureus. 2025;17(3):e81431. doi:10.7759/cureus.81431. PMID:40296953; PMCID:PMC12037203.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
Lisch W, Seitz B. The clinical landmarks of corneal dystrophies. Developments in ophthalmology. 2011;48:9-23. doi:10.1159/000324075. PMID:21540629.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern. San Francisco, CA: American Academy of Ophthalmology; 2024.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea. 2003;22(1):19-21.
Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969;1(4):314-324. PMID:4313418.
Kivelä T, Tarkkanen A, Frangione B, Ghiso J, Haltia M. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja’s syndrome. Invest Ophthalmol Vis Sci. 1994;35(10):3759-3769. PMID:8088963.
Steger B, Romano V, Biddolph S, Willoughby CE, Batterbury M, Kaye SB. Femtosecond laser-assisted lamellar keratectomy for corneal opacities secondary to anterior corneal dystrophies: an interventional case series. Cornea. 2016;35(1):6-13. PMID:26509759. doi:10.1097/ICO.0000000000000665.
Makale metnini kopyalayıp tercih ettiğiniz yapay zeka asistanına yapıştırabilirsiniz.
Makale panoya kopyalandı
Aşağıdaki yapay zeka asistanlarından birini açın ve kopyalanan metni sohbet kutusuna yapıştırın.