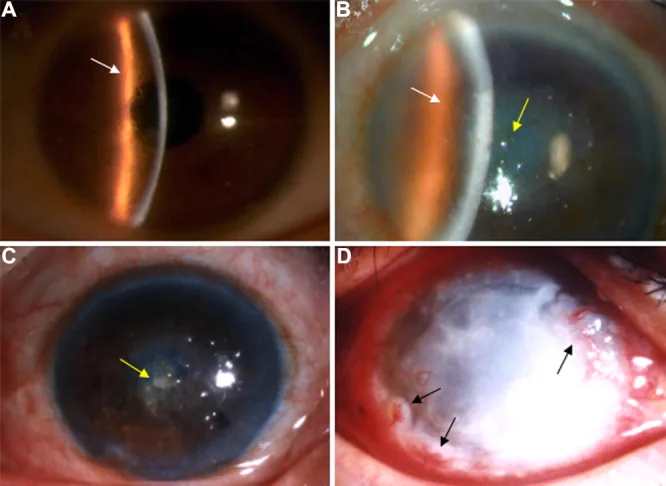
Die gittrige Hornhautdystrophie (lattice corneal dystrophy, LCD) ist eine erbliche Hornhautdystrophie, bei der sich Amyloid im Hornhautstroma ablagert und gitterartige lineare Trübungen verursacht. Sie wurde bereits in den 1890er Jahren beschrieben und wird in der klinisch-genetischen Klassifikation der IC3D (International Committee for Classification of Corneal Dystrophies) in der 2. Auflage als LCD1 und ihre Varianten (früher Typ 3, 3A, 1/3A, 4) zusammengefasst4).
LCD1, die körnige Hornhautdystrophie, die Reis-Bücklers-Hornhautdystrophie und die Thiel-Behnke-Hornhautdystrophie bilden eine Gruppe von Erkrankungen, die als „TGFBI-assoziierte Dystrophien“ bezeichnet werden. Das verantwortliche Gen TGFBI (transforming growth factor beta-induced gene) liegt auf dem langen Arm von Chromosom 5 (5q31) und wird autosomal-dominant vererbt. Das TGFBI-Protein (TGFBIp, Keratoepithelin, βig-h3) wird vom Hornhautepithel produziert und ist in der gesamten Hornhaut verteilt. Im Hornhautstroma ist es am Aufbau der Kollagenfasern beteiligt. Mutationen im selben Gen können je nach Ort und Art des Aminosäureaustauschs zu unterschiedlichen Ablagerungen (Hyalin oder Amyloid) und klinischen Bildern führen5).
Die repräsentative Mutation von LCD1 ist R124C, bei der Arginin an Position 124 im TGFBI-Gen durch Cystein ersetzt wird. Bei der Variante LCD IIIA wurden Mutationen wie L527R berichtet.
Das in der Hornhaut abgelagerte abnorme Protein färbt sich mit Kongorot rot und zeigt unter dem Polarisationsmikroskop eine charakteristische apfelgrüne Doppelbrechung, wodurch es als Amyloid identifiziert wird. Dieser Befund ist seit dem 19. Jahrhundert ein klassischer histologischer Diagnoseindikator für Amyloidose6).
Der früher als „gittrige Hornhautdystrophie Typ 2“ bezeichnete Typ ist eine Augenmanifestation der systemischen Gelsolin-Amyloidose (GSN-AMYL, Meretoja-Syndrom) und wird in der aktuellen IC3D-Klassifikation als „familiäre Amyloidose“ eingestuft, getrennt von der klassischen LCD behandelt4,10). Dieses 1969 von Meretoja in Finnland beschriebene Syndrom ist eine Erbkrankheit, die neben gittrigen Hornhauttrübungen auch progressive Hirnnervenlähmungen, Hauterschlaffung und systemische Symptome umfasst10,11). Da die Unterscheidung beider Erkrankungen im klinischen Alltag wichtig ist, werden beide in diesem Artikel aufgeführt.
Doppelkonturierte fadenförmige Trübungen im Pupillarbereich, rezidivierende Erosio corneae
LCD IIIA (Variante)
TGFBI (5q31)
L527R u. a.
Ab dem 40. Lebensjahr
Dicke, seilartige Gitterlinien im tiefen Stroma, keine Epithelschädigung
GSN-Typ (Meretoja)
GSN (9q34)
D187N, p.Glu580Lys2)
30–40 Jahre
Radiale Gitterlinien in der Peripherie, systemische Amyloidose
In Japan ist die häufigste TGFBI-assoziierte Dystrophie die granuläre Typ II (Avellino-Typ, R124H), während LCD1 im Vergleich dazu seltener ist. Da sich die Krankheitstypen jedoch durch nur wenige Basenpaare im selben TGFBI-Gen unterscheiden, ist bei überlappenden klinischen Bildern eine genetische Bestätigung wünschenswert. Die genaue Prävalenz von LCD insgesamt in Japan ist nicht berichtet, aber sie gehört zu den relativ seltenen Formen der Hornhautdystrophien.
QWie unterscheiden sich LCD1 und Meretoja-Syndrom?
A
LCD1 ist eine auf die Hornhaut beschränkte Amyloidablagerung aufgrund einer TGFBI-Genmutation, die im Alter von 10–20 Jahren im Pupillarbereich beginnt und häufig mit rezidivierenden Erosiones einhergeht. Das Meretoja-Syndrom (früher LCD2, GSN-Typ) hingegen ist eine Augenmanifestation einer systemischen Amyloidose aufgrund einer GSN- (Gelsolin-) Genmutation, die im Alter von 30–40 Jahren in der peripheren Hornhaut beginnt und die zentrale Transparenz lange erhält. Das Meretoja-Syndrom geht mit Hauterschlaffung, maskenartigem Gesicht, peripherer Neuropathie und Herzrhythmusstörungen einher2,10). In der IC3D-Klassifikation (2. Auflage) wird das Meretoja-Syndrom unabhängig von der gittrigen Hornhautdystrophie klassifiziert4).
Liu Z, et al. An R124C mutation in TGFBI caused lattice corneal dystrophy type I with a variable phenotype in three Chinese families. Mol Vis. 2008. Figure 2. PMCID: PMC2443752. License: CC BY.
Spaltlampenfoto: Verzweigte Gitterlinien und zentral betonte Trübungen im Hornhautstroma. Typisches klinisches Bild einer gittrigen Hornhautdystrophie.
Bei LCD1 sind viele Patienten im Kindesalter asymptomatisch; es zeigen sich nur feine Trübungen, die erst mit der retroilluminierenden Untersuchung am Spaltlampenmikroskop nachweisbar sind. Ab dem zweiten bis dritten Lebensjahrzehnt treten wiederkehrende Hornhauterosionen (rezidivierende Erosionen, RCE) auf, die sich durch plötzliche Augenschmerzen, Photophobie, Tränenfluss und Fremdkörpergefühl beim Aufwachen äußern. Etwa im Alter von 30 Jahren werden weiße Trübungen im vorderen Stroma der zentralen Hornhaut sichtbar, und ab dem vierten Lebensjahrzehnt schreitet die Sehverschlechterung fort.
Bei LCD IIIA (Variantform) treten in der Regel keine Epithelschäden auf; die Hauptbeschwerde ist eine langsame Sehverschlechterung nach dem 40. Lebensjahr.
Bei der früheren LCD2 (Meretoja-Syndrom) treten die Augensymptome im dritten bis vierten Lebensjahrzehnt auf, eine signifikante Sehbehinderung verzögert sich jedoch oft bis zum sechsten Lebensjahrzehnt11). Häufig treten systemische Symptome wie schlaffe Augenlider, maskenartiges Gesicht, progressive Hirnnervenausfälle und Herzrhythmusstörungen vor oder parallel auf2,10).
Die spaltlampenmikroskopischen Befunde der einzelnen Formen sind im Folgenden dargestellt.
LCD1 (klassische Form)
Erstmanifestation: Feine punktförmige und lineare Trübungen in der Bowman-Schicht bis zum vorderen Stroma im Pupillarbereich beider Augen.
Gitterlinien: Faden- oder linienförmige Trübungen mit Doppelkontur, die sich netz- oder sternförmig verflechten.
Fortgeschrittenes Stadium: Bildung einer eiförmigen oder runden, milchig-weißen Trübung in der zentralen Hornhaut.
Retroillumination: Feine, halbtransparente Gitterlinien, die bei direkter Beleuchtung schwer zu erkennen sind, werden deutlich sichtbar.
Fluorescein-Färbung: Aufgrund verminderter Epithelhaftung erscheint die Oberfläche rau.
Rezidivierende Erosionen: Treten häufig auf, da die Ablagerungen die basalen Epithelzellen und die Bowman-Membran betreffen.
LCD IIIA (Variantform)
Gitterlinien: Dicke, lange Gitterlinien im mittleren bis tiefen Stroma, manchmal mit dendritischen Verzweigungen. Auch bei direkter Beleuchtung sichtbar.
Phänotyp: Es gibt drei Muster: ① nur Gitterlinien, ② nur kleine körnige Ablagerungen, ③ eine Mischung aus beiden. Bei derselben Person können die beiden Augen unterschiedliche Phänotypen aufweisen, und es gibt auch einseitige Fälle.
Epithel: Normalerweise tritt keine Epithelschädigung auf.
Homozygot: Bei L527R-Homozygoten sind die Gitterlinien dicker und die zentralen körnigen Ablagerungen größer, aber der Unterschied ist nicht so ausgeprägt wie zwischen Hetero- und Homozygoten bei R124H (granulärer Typ II).
GSN-Typ (Meretoja)
Gitterlinien: Wenige, nicht sehr feine gitterförmige Ablagerungen, die radial von der Peripherie ausgehen.
Zentrale Transparenz: Die zentrale Transparenz bleibt lange nach Ausbruch der Erkrankung erhalten.
Erosionen des Epithels: Selten.
Systemische Befunde: Maskenhaftes Gesicht, hervorstehende Lippen mit Bewegungsstörungen, herabhängende Ohren, Schlupflider (Blepharochalasis) und andere Gesichtsveränderungen 2).
Bei LCD1 kann die zentrale runde Trübung je nach Fall besonders stark sein; es gibt einen Bericht über einen 56-jährigen R124C-Heterozygoten, bei dem aufgrund einer zentralen runden Trübung eine Hornhauttransplantation indiziert war.
QKann LCD1 auch bei Kindern diagnostiziert werden?
A
LCD1 im Kindesalter ist oft asymptomatisch, und Auffälligkeiten sind bei direkter Beleuchtung schwer zu finden. Mit der Durchleuchtungs- oder retroilluminationsmethode des Spaltlampenmikroskops können feine punktförmige bis lineare Trübungen im oberflächlichen zentralen Stroma nachgewiesen werden. Bei Kindern mit wiederkehrenden Hornhauterosionen sollte an LCD1 gedacht werden; eine Anamnese der Familienanamnese und eine Hornhautuntersuchung der Eltern werden empfohlen. Für die definitive Diagnose ist ein TGFBI-Gentest hilfreich.
Die ursächlichen Gene und repräsentativen Mutationen der gittrigen Hornhautdystrophie sind im Folgenden zusammengefasst.
TGFBI-assoziiert (LCD1, LCD IIIA, LCD IV)
Genlocus: 5q31 (TGFBI-Gen).
Vererbungsmodus: autosomal-dominant.
Häufigste LCD1-Mutation: R124C (Arg124Cys) ist die häufigste5).
Häufigste LCD-IIIA-Mutation: L527R (Leu527Arg) und andere wurden berichtet. Homozygote Fälle existieren ebenfalls.
De-novo-Mutation: Eine De-novo-Mutation TGFBI L509P wurde bei einem Fall mit LCD-IIIA-Phänotyp beschrieben1). Die Eltern hatten keine Mutation, aber eines der Kinder trug die Mutation1).
Rolle von TGFBIp: Wird vom Hornhautepithel produziert und in der gesamten Hornhaut verteilt; im Stroma ist es am Aufbau der Kollagenfasern beteiligt5).
GSN-assoziiert (Meretoja-Syndrom, früher LCD2)
Genlocus: 9q34 (GSN-Gen, Gelsolin).
Vererbungsmodus: autosomal-dominant.
Klassische Mutation: D187N (finnischer Typ) ist die häufigste; p.Asp187Tyr wurde ebenfalls beschrieben10,11).
Neue Mutation: p.Glu580Lys, berichtet in einer slowenischen Familie, liegt an der Grenze der G4-G5-Domäne und verursacht durch den Austausch von negativer zu positiver Ladung eine elektrostatische Abstoßung2).
Klinisches Bild: Neben gitterförmigen Hornhauttrübungen treten eine systemische Amyloidose mit Hauterschlaffung, Herzrhythmusstörungen, Nierenschädigung und Optikusneuropathie auf2).
Da es sich um eine Erbkrankheit handelt, ist die Familienanamnese der wichtigste Risikofaktor. Bei TGFBI können jedoch auch De-novo-Mutationen auftreten, sodass das Fehlen einer Familienanamnese die Erkrankung nicht ausschließt1). Der Vererbungsmodus ist autosomal-dominant; wenn ein Elternteil Mutationsträger ist, beträgt die Wahrscheinlichkeit einer Übertragung auf das Kind 50 %. Es gibt keine Geschlechterunterschiede, und auch ethnische Unterschiede sind bei LCD1 nicht deutlich, jedoch ist das Meretoja-Syndrom dafür bekannt, dass sich in Finnland viele betroffene Familien häufen11).
Der Beitrag von Umweltfaktoren ist nicht eindeutig; der Ausbruch und Verlauf dieser Erkrankung werden im Wesentlichen durch den Genotyp bestimmt. Allerdings kann die Häufigkeit rezidivierender Erosiones epithelialis durch trockene Umgebungen, Kontaktlinsentragen oder Traumata verstärkt werden. Refraktive Chirurgie (LASIK, SMILE usw.) kann eine rasche Verschlechterung TGFBI-assoziierter Dystrophien verursachen; daher ist bei der präoperativen Untersuchung von Patienten mit entsprechender Familienanamnese Vorsicht geboten5).
Zur Unterscheidung von LCD1, Variant-Typen und GSN-Typ werden Spaltlampenbefunde, histologische Befunde und genetische Befunde gemeinsam bewertet.
Klinische Untersuchungen
Spaltlampenmikroskopie: Bei direkter Beleuchtung können frühe gitterförmige Linien leicht übersehen werden. Mit der Durchleuchtungsmethode werden feine Trübungen vor dem Pupillenhintergrund erkannt, mit der retroilluminierenden Methode werden durchscheinende dünne gitterförmige Linien nachgewiesen.
Fluorescein-Färbung: Bei LCD1 ist die Epithelhaftung vermindert, was zu einer rauen Anfärbung führt. Sie ist auch zur Beurteilung des Ausmaßes von Erosionen nützlich.
Konfokale Mikroskopie der Hornhaut: Ablagerungen im Stroma können auf zellulärer Ebene beobachtet werden.
Definitive Diagnose
Gentest: Der Nachweis von Mutationen im TGFBI-Gen und GSN-Gen bestätigt den Krankheitstyp. Da unterschiedliche Mutationen trotz gleichen Phänotyps die Geschwindigkeit von Rezidiv und Progression beeinflussen, hat dies direkte Auswirkungen auf den Behandlungsplan.
Pathologische Untersuchung: Die Kongorot-Färbung zeigt eine rote Farbe, und unter dem Polarisationsmikroskop zeigt sich eine apfelgrüne Doppelbrechung, wodurch Amyloid bestätigt wird6).
Immunhistochemie: Die Unterscheidung der Krankheitstypen ist mit Anti-TGFBIp-Antikörpern und Anti-Gelsolin-Antikörpern möglich.
Erhebung der Familienanamnese: Aufgrund des autosomal-dominanten Erbgangs unterstützt die Untersuchung der Hornhautbefunde bei Eltern und Geschwistern die Diagnose.
Granuläre Hornhautdystrophie Typ II (Avellino-Typ, TGFBI R124H): Dies ist die häufigste TGFBI-assoziierte Dystrophie in Japan und zeigt ein Mischbild aus granulären Ablagerungen und gitterförmigen Linien. Zur Unterscheidung von LCD1 ist ein Gentest zuverlässig.
Sekundäre Hornhautamyloidose: Nicht erblich, Amyloid lagert sich sekundär als Folge chronischer Augenoberflächenreizung wie Trichiasis oder Keratokonus ab. Fehlende Familienanamnese und Vorliegen einer Grunderkrankung sind Unterscheidungsmerkmale.
Fleckige Hornhautdystrophie: Autosomal-rezessive Vererbung durch CHST6-Genmutation, mit diffuser Mattglas-Trübung und Endothelanomalien.
Gelatinöse tropfenförmige Hornhautdystrophie: Autosomal-rezessive Vererbung durch TACSTD2-Genmutation, zeigt milchig-weiße gelatinöse Erhebungen. In Japan relativ häufig.
QWarum ist die genetische Untersuchung wichtig?
A
Obwohl die gittrige Hornhautdystrophie phänotypisch ähnlich ist, unterscheiden sich Progressionsgeschwindigkeit, Rezidivhäufigkeit, Behandlungsoptionen und das Vorhandensein systemischer Komplikationen erheblich, je nach verantwortlichem Gen und Mutationsort. Bei LCD1 mit TGFBI-Mutation und Meretoja-Syndrom mit GSN-Mutation unterscheiden sich Behandlungsstrategie und Notwendigkeit systemischer Abklärungen grundlegend2,10). Zudem wurden Fälle mit de novo-Mutationen berichtet, bei denen die Familienanamnese allein den Subtyp nicht bestimmen kann1), weshalb die genetische Untersuchung für die definitive Diagnose und Klassifikation unerlässlich ist.
Im Kindes- bis jungen Erwachsenenalter, wenn keine Symptome oder nur feine Trübungen vorliegen, wird abgewartet. Die Progression wird alle sechs Monate bis ein Jahr mittels Spaltlampenuntersuchung beurteilt.
Bei rezidivierenden Erosionen, einem Kernsymptom der LCD1, ist die folgende konservative Therapie der erste Schritt.
Akutbehandlung: Kontinuierliches Tragen von therapeutischen weichen Kontaktlinsen zum Schutz des Hornhautepithels. Kombination mit antibakteriellen Augentropfen zur Sekundärinfektionsprophylaxe. Auftragen von Augensalbe zur Befeuchtung und zum Epithelschutz.
Rezidivprophylaxe: Die Anwendung von Augensalbe vor dem Schlafengehen kann das Wiederauftreten von RCE-Anfällen reduzieren. In trockener Umgebung tagsüber auch künstliche Tränen oder Gleitmittel verwenden.
Bei LCD1, bei dem die Amyloidablagerungen hauptsächlich in der Hornhautoberfläche liegen, ist bei starker zentraler Trübung oder wiederholten rezidivierenden Hornhauterosionen die phototherapeutische Keratektomie (PTK) mit dem Excimerlaser die erste Wahl7,8). In der Regel tritt kein frühes Rezidiv auf, aber im Laufe der Zeit ist ein Rezidiv unvermeidbar; eine PTK-Behandlung kann am selben Auge bis zu etwa zweimal durchgeführt werden.
Bei Heterozygoten ist das Wiederauftreten langsam und eine erneute Behandlung ist selten erforderlich. Bei Homozygoten besteht eine Tendenz zu früherem Wiederauftreten im Vergleich zu Heterozygoten. Die Rezidivrate nach PTK steigt wie bei anderen TGFBI-assoziierten Dystrophien im Laufe der Zeit an, und bei längerer Nachbeobachtung werden bei vielen Fällen gewisse Rezidivbefunde festgestellt8).
Als Fall, der die Wirksamkeit von PTK zeigt, wurde bei einem Fall von LCD IIIA aufgrund einer de novo Mutation von TGFBI L509P eine FD-OCT-geführte PTK von 60 µm durchgeführt, und der bestkorrigierte Visus (BCVA) verbesserte sich von 20/400 auf 20/501). 45 Monate nach dem Eingriff wurde keine Sehverschlechterung oder signifikantes Wiederauftreten festgestellt1).
Laut dem Preferred Practice Pattern der AAO zu Hornhautödem und -trübung ist PTK bei granulären und gittrigen Hornhautdystrophien eine „vernünftige Option (reasonable means)“, die den Übergang zu DALK oder perforierender Keratoplastik verzögern kann, jedoch mit einem Risiko für postoperativen Haze verbunden ist. Bei wiederholter Durchführung wird die Kombination mit Mitomycin C als Mittel zur Unterdrückung rezidivierender Narben und Stromaablagerungen in Betracht gezogen. Es wird gewarnt, dass das Risiko einer Hornhautektasie steigt, wenn die Ablation das vordere Drittel des Stromas überschreitet oder das verbleibende Bett weniger als 250 µm beträgt7).
Bei wiederholtem Wiederauftreten oder wenn die Trübung tiefere Stromaschichten betrifft, wird eine Hornhauttransplantation gewählt. Bei LCD1 ist eine Hornhauttransplantation in der Regel erst nach dem 40. Lebensjahr indiziert. Da die Hornhautendothelzellen bei LCD in der Regel normal sind, wird das Verfahren je nach Tiefe der Trübung ausgewählt.
Hohe Sehkraftwiederherstellung, aber Risiko von Abstoßung und Rezidiv
In den letzten Jahren wird die DALK aufgrund des geringeren Abstoßungsrisikos und der mit der perforierenden Keratoplastik vergleichbaren Sehergebnisse zunehmend als neue Erstlinientherapie eingesetzt.
Ein Rezidiv der LCD nach Keratoplastik ist unvermeidlich. Die Rezidivrate nach perforierender Keratoplastik wird mit 17,8 % nach 5 Jahren, 26 % nach 8 Jahren und 56 % nach 15 Jahren angegeben9). Da die rezidivierende Trübung meist auf die oberflächlichen Schichten beschränkt ist, kann sie mittels PTK entfernt und die Zeit bis zur erneuten Transplantation verlängert werden. Bei LCD IIIA (Variante) ist oft keine Behandlung erforderlich, solange die Sehkraft nicht stark beeinträchtigt ist.
QWie wirksam ist die PTK?
A
Die PTK kann oberflächliche Amyloidablagerungen effektiv entfernen und führt zu einer Verbesserung der Sehkraft sowie einer Verringerung rezidivierender Erosionen. Bei einem Fall von LCD IIIA verbesserte sich der bestkorrigierte Visus nach einer PTK von 60 µm von 20/400 auf 20/50, und es trat 45 Monate lang kein Rezidiv auf1). Bei Heterozygoten ist das Rezidiv langsam, bei Homozygoten tritt es früh auf. Tiefe Läsionen können mit PTK nicht entfernt werden, daher ist bei tiefer Trübung eine DALK oder perforierende Keratoplastik erforderlich7).
QTritt nach einer Hornhauttransplantation ein Rezidiv auf?
A
Ein Rezidiv der LCD nach Keratoplastik ist unvermeidlich. Die Rezidivrate nach perforierender Keratoplastik wird mit 17,8 % nach 5 Jahren, 26 % nach 8 Jahren und 56 % nach 15 Jahren angegeben9). Da die rezidivierende Trübung jedoch meist auf die oberflächlichen Schichten des Transplantats beschränkt ist, kann sie mittels PTK entfernt und die Lebensdauer des Transplantats verlängert werden. Die tiefe lamelläre Keratoplastik (DALK) hat im Vergleich zur perforierenden Keratoplastik ein geringeres Risiko einer endothelialen Abstoßung und wird als neue Erstlinientherapie angesehen7).
Im Zentrum der Pathologie der LCD1 steht die abnorme Akkumulation von TGFBIp (Kerato-Epithelin, βig-h3). TGFBIp ist ein Strukturprotein, das normalerweise vom Hornhautepithel produziert wird, in der gesamten Hornhaut verteilt ist und im Stroma am Aufbau von Kollagenfasern und an der Zelladhäsion beteiligt ist 5). Das durch die R124C-Mutation produzierte abnorme Protein unterliegt einer Fehlfaltung und Selbstaggregation und lagert sich als unlösliche Amyloidfibrillen in der Bowman-Schicht und im oberflächlichen Stroma ab. Im fortgeschrittenen Stadium breitet sich die Ablagerung in die tiefen Stromaschichten aus.
Die Amyloidablagerung führt zu Veränderungen der epithelialen Adhäsionsstrukturen der vorderen Hornhaut und verursacht eine Degeneration der epithelialen Basalzellen sowie eine Degeneration der Epithelschicht mit Defekten der Bowman-Membran. Diese strukturelle Störung bildet die pathologische Grundlage für die rezidivierende Hornhauterosion.
Beim TGFBI-Gen bestimmen die Mutationsstelle und der ausgetauschte Aminosäurerest das klinische Bild. R124C verursacht LCD1, R124H die granuläre Hornhautdystrophie Typ II (Avellino-Typ) und R124L die Reis-Bücklers-Hornhautdystrophie5). Der molekulare Mechanismus, durch den bereits ein einzelner Aminosäureaustausch das Ablagerungsmaterial (Amyloid vs. Hyalin vs. beides) und die Ablagerungsstelle bestimmt, ist noch nicht vollständig aufgeklärt, aber es wird angenommen, dass die Zugehörigkeit der Mutationsstelle zu einer bestimmten Domäne von βig-h3 und der Einfluss auf die Faltungsstabilität entscheidend sind.
Bei LCD IIIA führen tiefe Mutationen wie L527R zu dicken, seilartigen gitterförmigen Linien und einem späten Krankheitsbeginn ohne Epithelschädigung. Die schichtenspezifische Lokalisation der Ablagerungen kann durch den Sekretions- und Diffusionsgradienten von βig-h3 von den produzierenden Zellen (epitheliale Basalzellen) in das Stroma sowie durch Unterschiede in der Faltungsstabilität des mutierten Proteins erklärt werden. Es wird angenommen, dass R124C bevorzugt den Weg von Faltungsintermediaten zur Amyloidfibrillenbildung einschlägt und Amyloid in der Umgebung der Bowman-Schicht akkumuliert 5). Die L527R-Mutation hingegen bildet ein relativ stabiles fehlgefaltetes Protein, das sich langsam in den tieferen Stromaschichten ablagert.
Hinteres Hornhautamyloid und Risiko intraokularer Chirurgie
Bisher wurde angenommen, dass die Amyloidablagerung bei LCD1 auf die vordere Hornhaut (Bowman-Schicht bis oberflächliches Stroma) beschränkt ist. Neuere pathologische Untersuchungen haben jedoch gezeigt, dass auch in der hinteren Hornhaut in der Nähe der Descemet-Membran Amyloidablagerungen vorhanden sind 3). Die Amyloidablagerung in der hinteren Hornhaut könnte die Adhäsion der Descemet-Membran beeinträchtigen und zur Descemet-Membran-Ablösung während der Kataraktchirurgie beitragen 3). Es wird vermutet, dass ein ähnlicher Mechanismus, wie er bei der vorderen Hornhaut die Epitheladhäsion stört, auch im hinteren Bereich wirkt 3).
Gelsolin, das verantwortliche Molekül für die frühere LCD2 (Meretoja-Syndrom), kommt sowohl im Zytoplasma als auch extrazellulär vor und ist ein Protein, das über Aktinbindung an Zellbewegung, Zellteilung und Apoptose beteiligt ist. Die klassische D187N-Mutation, auch finnischer Typ genannt, führt zu einem Phänotyp, der hauptsächlich durch gitterförmige Hornhautablagerungen und Hirnnervenstörungen gekennzeichnet ist 11). Die in einer slowenischen Familie beschriebene neue p.Glu580Lys-Mutation liegt an der Grenze der G4-G5-Domänen und führt durch den Austausch von negativ geladenem Glutamat gegen positiv geladenes Lysin zu einer elektrostatischen Abstoßung, was die Verbindung und Stabilität zwischen den Domänen verringert 2). Mutiertes Gelsolin wird im Plasma durch Furin und MT1-MMP abnormal gespalten und setzt 8 kDa und 5 kDa große Amyloid-Vorläuferfragmente frei. Diese lagern sich im Hornhautstroma, der Haut, der Gefäßwand, peripheren Nerven und den Nierenglomeruli ab und verursachen die für das Meretoja-Syndrom charakteristischen Multiorgansymptome 2,11). Hornhautablagerungen treten oft vor anderen systemischen Symptomen auf und können Augenärzten einen ersten Hinweis auf die Erkrankung geben.
Es wurde über das Auftreten von LCD durch De-novo-Mutationen im TGFBI-Gen berichtet 1). Auch bei Fällen ohne Familienanamnese sollte die Möglichkeit einer De-novo-Mutation in Betracht gezogen werden, und eine Bestätigung durch Gentests wird empfohlen 1). Die L509P-Mutation ist selten, zeigt aber ein breites phänotypisches Spektrum von Reis-Bücklers-Hornhautdystrophie-ähnlich bis LCD-IIIA-ähnlich 1).
Beim GSN-Gen wurde zusätzlich zu den herkömmlichen p.Asp187Asn/Tyr-Mutationen eine neue p.Glu580Lys-Mutation beschrieben, die mit gitterförmiger Hornhautdystrophie, Hauterschlaffung, Herzrhythmusstörungen, Nierenschädigung und Optikusneuropathie assoziiert ist und eine systemische Amyloidose verursacht 2).
Hintere Hornhautläsionen und Management intraokularer Chirurgie
Bei LCD1-Patienten wurde pathologisch gezeigt, dass Amyloidablagerungen in der hinteren Hornhaut vorhanden sind und die Adhäsion der Descemet-Membran beeinträchtigen können 3). Bei intraokularen Eingriffen wie Kataraktoperationen ist auf das Risiko einer Descemet-Membran-Ablösung zu achten.
Diese Erkenntnis hat klinische Implikationen für die Beurteilung der Kataraktoperationsindikation und die Operationsplanung bei LCD1-Patienten.
Die Entwicklung präziserer Operationsverfahren wie der femtosekundenlaser-assistierten lamellären Keratektomie (FLK) und der femtosekundenlaser-assistierten lamellären Keratoplastik (FALK) schreitet voran 12). Diese werden aufgrund der verbesserten Glätte der Schnittfläche und der reproduzierbaren Tiefenkontrolle zunehmend als ergänzende Option zur konventionellen PTK betrachtet.
Da TGFBI-Mutationen autosomal-dominante Gain-of-Function-Mutationen sind, werden mutationsallelspezifische siRNAs, Antisense-Oligonukleotide und allelspezifisches Knockout mittels CRISPR-Cas9 in präklinischen Studien untersucht. Die Hornhaut ist aufgrund der Möglichkeit lokaler Verabreichung und ihrer Immunprivilegien ein vorteilhaftes Zielorgan für die Gentherapie. Derzeit gibt es jedoch keine klinische Anwendung, und alle Ansätze erfordern weitere Überprüfungen der langfristigen Sicherheit und Wirksamkeit.
Niedermolekulare Verbindungen, die auf den Aggregationsprozess von TGFBIp und mutiertem Gelsolin abzielen, molekulare Chaperone (wie Hsp70-Induktoren) und Amyloidfibrillen-Bindungshemmer werden in der Grundlagenforschung untersucht. Für die systemische Gelsolin-Amyloidose werden Medikamente, die die Spaltung von mutiertem Gelsolin im Plasma hemmen, in einigen präklinischen Studien evaluiert 2). Zukünftig werden solche molekular gezielten Therapien als grundlegende Behandlungen erwartet, die die konventionelle physikalische Entfernung (PTK, Hornhauttransplantation) ersetzen könnten.
Massenspektrometrische Proteomanalysen der Hornhaut haben gezeigt, dass in den Ablagerungen der LCD1 nicht nur TGFBIp, sondern auch mehrere abnormale Proteine gemeinsam ausgefällt werden können. Für die zukünftige klinische Anwendung wird der Beitrag dieser kopräzipitierten Proteine zur Pathogenese untersucht.
Ji YW, Ahn H, Shin KJ, Kim TI, Seo KY, Stulting RD, et al. De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. Journal of clinical medicine. 2022;11(11). doi:10.3390/jcm11113055. PMID:35683443; PMCID:PMC9181583.
Potrc M, Volk M, de Rosa M, Pizem J, Teran N, Jaklic H, Maver A, Drnovsek-Olup B, Bollati M, Vogelnik K, Hocevar A, Gornik A, Pfeifer V, Peterlin B, Hawlina M, Fakin A. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. Int J Mol Sci. 2021;22(3):1084.
Matsumoto A, Fukuoka H, Sotozono C. Descemet’s Membrane Detachment During Cataract Surgery in Lattice Corneal Dystrophy Type I: Histopathological Analysis of Posterior Corneal Involvement. Cureus. 2025;17(3):e81431. doi:10.7759/cureus.81431. PMID:40296953; PMCID:PMC12037203.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
Lisch W, Seitz B. The clinical landmarks of corneal dystrophies. Developments in ophthalmology. 2011;48:9-23. doi:10.1159/000324075. PMID:21540629.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern. San Francisco, CA: American Academy of Ophthalmology; 2024.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea. 2003;22(1):19-21.
Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969;1(4):314-324. PMID:4313418.
Kivelä T, Tarkkanen A, Frangione B, Ghiso J, Haltia M. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja’s syndrome. Invest Ophthalmol Vis Sci. 1994;35(10):3759-3769. PMID:8088963.
Steger B, Romano V, Biddolph S, Willoughby CE, Batterbury M, Kaye SB. Femtosecond laser-assisted lamellar keratectomy for corneal opacities secondary to anterior corneal dystrophies: an interventional case series. Cornea. 2016;35(1):6-13. PMID:26509759. doi:10.1097/ICO.0000000000000665.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.