Die gelatinöse tropfenförmige Hornhautdystrophie (gelatinous drop-like corneal dystrophy: GDLD) ist eine erbliche Hornhauterkrankung, bei der sich Amyloid unter dem Hornhautepithel ablagert und zu einer beidseitigen deutlichen Sehverschlechterung führt.
Diese Erkrankung wurde erstmals 1914 von Nakaizumi beschrieben, und seit Kiyosawa sie 1932 als „gelatinöse tropfenförmige Hornhautdegeneration“ benannte, wird sie unter diesem Namen geführt. In der IC3D-Klassifikation (International Committee for Classification of Corneal Dystrophies) wird sie als epitheliale Dystrophie mit der Abkürzung GDLD eingestuft.
Das verantwortliche Gen, TACSTD2 (tumor-associated calcium signal transducer 2), wurde 1999 von Tsujikawa et al. identifiziert. Es handelt sich um ein Single-Exon-Gen auf Chromosom 1p324).
Prävalenz: Weltweit selten, aber in Japan als relativ häufige Erkrankung berichtet 1). Es wird angenommen, dass die Inzidenz aufgrund des Rückgangs von Blutsverwandtenehen abgenommen hat 2)
Regionale Unterschiede: In Japan relativ häufig, in Europa und Nordamerika kaum berichtet
Q118X-Mutation: Gründermutation bei japanischen Patienten, die über 80 % der krankheitsverursachenden Chromosomen ausmacht 2)
Erkrankungsalter: Tritt meist vor dem 20. Lebensjahr auf
Im Jahr 2019 wurde sie als spezifische seltene Krankheit „Gelatinöse tropfenförmige Hornhautdystrophie“ anerkannt und für die Kostenübernahme von medizinischen Behandlungen qualifiziert 2). Im Rahmen des Forschungsprojekts des Ministeriums für Gesundheit, Arbeit und Soziales zu refraktären Krankheiten wurden Diagnosekriterien und Schweregradeinteilungen entwickelt 2).
QTritt GDLD auch außerhalb Japans auf?
A
GDLD wurde weltweit berichtet, ist aber in Japan häufiger. In Europa und Nordamerika gibt es fast keine Fälle. Für das TACSTD2-Gen wurden über 20 Mutationen berichtet, was auf genetische Heterogenität hinweist. In Japan ist die auf 1p32 lokalisierte Q118X-Mutation als Gründermutation häufig und macht über 80 % der krankheitsverursachenden Chromosomen bei japanischen Patienten aus.
Tritt meist vor dem 20. Lebensjahr auf. Die folgenden Symptome treten bereits in der Kindheit auf.
Photophobie (Lichtempfindlichkeit): Bereits früh ein auffälliges Symptom
Fremdkörpergefühl: Aufgrund der gallertartigen Erhebungen auf der Hornhautoberfläche
Tränenfluss: Begleitend zu Reizsymptomen
Sehverschlechterung: Nimmt mit fortschreitender Amyloidablagerung allmählich ab. Im Erwachsenenalter wird sie deutlich
Mit zunehmendem Alter nehmen Anzahl und Größe der Amyloidablagerungen zu. Sie werden zu grau-weißen bis gelben Ablagerungen und bedecken schließlich den größten Teil der Hornhaut, insbesondere im Bereich der Lidspalte 2). Es kommt zu Gefäßeinsprossung von der Peripherie, deutlicher Sehverschlechterung und Augenschmerzen, hinzu kommen kosmetische Probleme, was die Lebensqualität des Patienten erheblich beeinträchtigt.
Klinische Befunde (vom Arzt bei der Untersuchung festgestellte Befunde)
Die Hornhauttrübung wird je nach Morphologie in vier Typen eingeteilt. Diese können mittels Spaltlampenmikroskopie des vorderen Augenabschnitts unterschieden werden 2,3).
Maulbeerartig
typical mulberry type: Der typischste Typ.
Zentraler Bereich der Hornhaut: Grau-weiße erhabene Läsionen häufen sich und ähneln einer Maulbeere.
Subepitheliales Amyloid: Milchig-weiße, durchscheinende, gallertartige Erhebungen nehmen von der Mitte zur Peripherie hin zu.
Bandförmige Keratopathie
band-keratopathy type: Kann im Frühstadium auftreten.
Lidspaltenbereich: Oberflächliche Trübung. Ähnelt der bandförmigen Keratopathie.
Bindehautläsionen: Auch die Bindehaut kann betroffen sein.
Kumquatartig
kumquat-like type: Häufig bei fortgeschrittenen Fällen.
Diffuse gelb-weiße Ablagerungen: Die gesamte Hornhaut verfärbt sich gelb und ähnelt einer Kumquat.
Gefäßeinsprossung: Kann von oberflächlicher Neovaskularisation begleitet sein.
Ausbreitung auf das Stroma : Die Läsion erstreckt sich auf das Hornhautstroma.
Gefäßeinsprossung : Milchig-weiße bis gelbe gallertartige Erhebungen mit Gefäßeinsprossung.
Ide et al. berichteten über das detaillierte klinische Spektrum von 34 japanischen Fällen und zeigten, dass trotz derselben TACSTD2-Genmutation (Q118X-Homozygot) vier Phänotypen nebeneinander existieren3).
Weitere charakteristische Befunde sind:
Verzögerte Fluorescein-Anfärbung (delayed staining) : Trotz fehlender Hornhautepithelschädigung wird aufgrund erhöhter Permeabilität durch unvollständige Tight-Junction-Bildung einige Minuten nach Fluorescein-Gabe Fluoreszenz beobachtet2)
Epithelverdünnung : Das Hornhautepithel ist im Bereich der gallertartigen Erhebungen verdünnt
Gefäßeinsprossung : Oberflächliche Gefäßeinsprossung in der peripheren Hornhaut
Durch die Anomalie des TACSTD2-Gens geht die normale intrazelluläre Lokalisation von Claudin-1 und Claudin-7, den Strukturproteinen der Tight Junctions im Hornhautepithel, verloren, und die Barrierefunktion des Epithels wird beeinträchtigt5). Infolgedessen dringen Proteine wie Laktoferrin aus der Tränenflüssigkeit in die Hornhaut ein, bilden Amyloidfibrillen und lagern sich unter dem Epithel ab. Nakatsuka et al. zeigten durch molekularbiologische Analyse japanischer Familien, dass TACSTD2 für die normale Claudin-Lokalisation essentiell ist, und stellten klar, dass die Pathologie der GDLD auf eine Funktionsstörung der Tight Junctions zurückzuführen ist5).
Familienanamnese : Aufgrund des autosomal-rezessiven Erbgangs besteht ein Risiko, die Krankheit zu entwickeln, wenn beide Elternteile Träger sind.
Japanische Abstammung: Die Q118X-Nonsense-Mutation ist eine Gründermutation in Japan und macht über 80 % der krankheitsverursachenden Chromosomen aus2)
Blutsverwandtschaft: Die Eltern des Indexpatienten sind häufig blutsverwandt. Allerdings kann die Krankheit auch bei compound-heterozygoten Personen auftreten, die aus Ehen zwischen nicht verwandten Familien stammen.
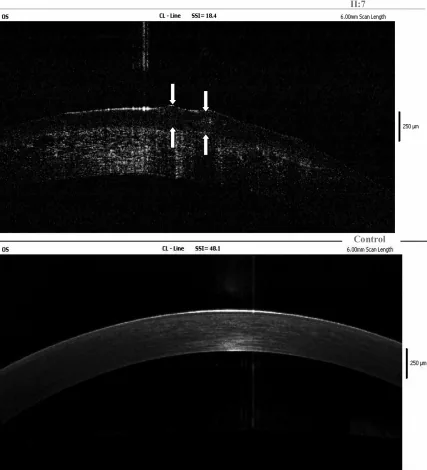
Yang Jing, Chun Liu, Liya Wang A novel TACSTD2 mutation identified in two Chinese brothers with gelatinous drop-like corneal dystrophy 2009 Aug 14 Mol Vis. 2009 Aug 14; 15:1580-1588 Figure 5. PMCID: PMC2728569. License: CC BY.
Die Fourier-Domänen-OCT-Aufnahme der linken Hornhaut des Indexpatienten zeigt Amyloidablagerungen im Hornhautepithel und im oberflächlichen Stroma; die Pfeile zeigen die Position der gelatinösen tropfenartigen Läsionen an. Dies entspricht den Amyloidablagerungen, die im Abschnitt „4. Diagnose und Untersuchungsmethoden“ behandelt werden.
Diagnosekriterien (Forschungsgruppe des Gesundheitsministeriums)
Die Diagnosekriterien für GDLD wurden von der Forschungsgruppe des Ministeriums für Gesundheit, Arbeit und Soziales im Rahmen des Projekts „Erstellung und Verbreitung von Behandlungsleitlinien für seltene Erkrankungen des vorderen Augenabschnitts“ festgelegt2). Wenn nach diesen Kriterien die Diagnose „Definite“ gestellt wird, gilt die Erkrankung als designierte seltene Krankheit.
A. Symptome (mindestens eines der folgenden)
Verminderte Sehschärfe
Lichtempfindlichkeit
Fremdkörpergefühl
Tränenfluss
B. Untersuchungsbefunde
Es zeigen sich grau-weiße, erhabene Ansammlungen von Amyloidablagerungen (maulbeerartig) direkt unter dem Hornhautepithel im zentralen Bereich der Hornhaut und der Lidspalte beider Augen.
Es zeigt sich eine verzögerte Anfärbung (delayed staining): Die Fluoreszenz wird einige Minuten nach der Fluorescein-Färbung beobachtet, obwohl keine Hornhautepithelschädigung vorliegt.
Es zeigt sich eine oberflächliche Gefäßeinsprossung im peripheren Bereich der Hornhaut.
C. Differentialdiagnose : Ausschluss einer sekundären (erworbenen) Hornhautamyloidose und einer klimatischen Tröpfchenkeratopathie.
D. Extraokuläre Komplikationen : Keine.
E. Genetische Untersuchung : Nachweis einer Anomalie im TACSTD2-Gen.
Die Bedingungen für die Kategorie Definitiv sind, wenn eine der folgenden Voraussetzungen erfüllt ist 2):
Fälle, die Kriterium D erfüllen, eines der Kriterien A aufweisen, Kriterium B1 erfüllen und die in C zu differenzierenden Erkrankungen ausschließen können.
Fälle, die Kriterium D erfüllen, eines der Kriterien A aufweisen, Kriterium B2 oder B3 erfüllen, Kriterium E erfüllen und die in C zu differenzierenden Erkrankungen ausschließen können.
B1 (maulbeerartige Ablagerungen) ist ein sehr charakteristischer Befund; in typischen Fällen bereitet die Diagnose keine Schwierigkeiten. In atypischen Fällen wird die Diagnose durch Kombination von A bis C mit der genetischen Untersuchung (E) gestellt 2).
Schweregradeinteilung (gemäß der Bekanntmachung zu designierten seltenen Erkrankungen)
Der Schweregrad wird basierend auf dem bestkorrigierten Visus des besseren Auges in die Grade I bis IV eingeteilt 2).
Schweregrad
Kriterium
Kostenübernahme
Grad I
Nur ein Auge betroffen, das andere Auge gesund
×
Grad II
Beide Augen betroffen, korrigierte Sehschärfe des besseren Auges ≥ 0,3
×
Grad III
Beide Augen betroffen, korrigierte Sehschärfe des besseren Auges ≥ 0,1 und < 0,3
○
Grad IV
Beide Augen betroffen, korrigierte Sehschärfe des besseren Auges < 0,1
○
Bei Diagnose als Definite wird die Erkrankung als designierte seltene Krankheit anerkannt, und ab Schweregrad III kann eine Kostenübernahme für medizinische Behandlungen beantragt werden 2). Wenn aufgrund von Sekundärglaukom etc. eine Gesichtsfeldeinschränkung (zentrales Restgesichtsfeld ≤ 20° mit Goldmann I/4-Marke) am besseren Auge vorliegt, erhöht sich der Schweregrad um eine Stufe.
Spaltlampenmikroskopie: Beobachtung der gräulich-weißen erhabenen Läsionen von der Hornhautmitte bis zur Lidspaltenregion, Unterscheidung der 4 morphologischen Typen (maulbeerartig, bandförmig, kumquatartig, stromale Trübung).
Fluorescein-Permeabilitätstest (verzögerte Anfärbung) : Aufgrund einer Funktionsstörung der Tight Junctions dringt der Farbstoff schnell in das Hornhautgewebe ein 2)
TACSTD2-Gentest : Seit dem Geschäftsjahr 2020 als Hornhautdystrophie-Gentest (D006-20) von der Krankenkasse erstattet 2). TACSTD2 ist ein Single-Exon-Gen, was die Suche erleichtert. Besonders nützlich für die Diagnose atypischer Fälle
Gewebeuntersuchung (Hornhautschnitt) : Färbung mit Kongorot ergibt eine orange-rote Farbe, und unter dem Polarisationsmikroskop zeigt sich eine apfelgrüne Doppelbrechung, die Amyloid bestätigt
Sekundäre Hornhautamyloidose : Amyloidablagerung durch chronische Reizung wie Trichiasis, Entropium, Keratokonusvorwölbung, Tragen harter Kontaktlinsen. Fehlende Familienanamnese und Hintergrund einer chronischen Augenoberflächenentzündung sind Unterscheidungsmerkmale. Kann gelatineartige Erhebungen oder gitterartige Erscheinungen aufweisen; zur Bestätigung ist eine Gewebeuntersuchung erforderlich
Klimatische Tröpfchenkeratopathie : Häufig bei Männern über 40 Jahren. Tritt in Wüsten- oder extrem kalten Regionen auf, verursacht durch UV-Strahlung und Trockenheit. Zeigt gelbe bis grau-weiße erhabene Hornhautläsionen
Bandförmige Hornhautdegeneration : Kalziumsalze lagern sich subepithelial ab. Beginnt in der Peripherie bei 3 und 9 Uhr und schreitet zur Mitte fort
Gitterförmige Hornhautdystrophie Typ I : TGFBI-Genmutation R124C, autosomal-dominante Vererbung. Zeigt verzweigte faserige Trübungen im Hornhautstroma
QWird der Gentest von der Krankenkasse übernommen?
A
Der TACSTD2-Gentest wird seit 2020 als „Hornhautdystrophie-Gentest (D006-20)“ von der Krankenkasse erstattet. Allerdings muss die Einrichtung eine Zertifizierung erhalten, nachdem sie ein System zur Durchführung des Tests innerhalb der Einrichtung eingerichtet hat. Da TACSTD2 ein Single-Exon-Gen ist, das leicht zu durchsuchen ist, und mehr als 80 % der japanischen Patienten die Gründer-Mutation Q118X aufweisen, ist dieser Test besonders nützlich für die Diagnose atypischer Fälle 2).
Die Behandlung der GDLD wird je nach Ausdehnung der Trübung und dem Grad der Sehbehinderung ausgewählt. Da es sich um eine Erbkrankheit handelt, ist die Rezidivrate bei jeder Therapie extrem hoch, was die größte Herausforderung darstellt 2). Fälle von Erblindung aufgrund von Komplikationen mehrfacher Hornhauttransplantationen oder sekundärem Glaukom sind nicht selten.
Künstliche Tränen : werden als symptomatische Therapie zur Linderung von Oberflächenreizungen eingesetzt.
Kontinuierliches Tragen von therapeutischen weichen Kontaktlinsen (SCL) : kann das Wiederauftreten von gelatinösen Erhebungen unterdrücken und den Operationsabstand verlängern.
Das kontinuierliche Tragen von therapeutischen SCL wird als konservative und adjuvante Behandlung in Betracht gezogen 6). Maeno et al. zeigten 2020 in einer prospektiven Beobachtungsstudie an GDLD-Patienten, dass das Tragen von therapeutischen SCL das Wiederauftreten von grauweißen bis gelben gelatinösen Erhebungen signifikant unterdrückt 7). Es wird auch zur Vorbeugung von Rezidiven nach der Operation empfohlen.
Rezidivrate : Nach perforierender Keratoplastik ist das Rezidiv mit 97 % innerhalb von 4 Jahren hoch. Die DALK hat den Vorteil des Endothelperhalts.
Limbustransplantation
Limbale Stammzelltransplantation, korneale Epithelplastik : in Kombination mit Hornhauttransplantation.
Ziel : Die Augenoberfläche mit dem Korneaepithel des Transplantats bedecken und ein erneutes Einwachsen des Wirtsepithels verhindern 10,11).
Mehrere Berichte aus Japan liegen zu den Langzeitergebnissen der PTK vor. Ōura et al. zeigten die Langzeitergebnisse der PTK bei GDLD-Patienten und berichteten über die Nützlichkeit zur Verlängerung des rezidivfreien Intervalls 8). Hieda et al. analysierten in einer multizentrischen japanischen Studie den Zeitpunkt des Rezidivs und die klinischen Ergebnisse nach PTK detailliert 9).
Die Kombination mit einer limbalen Stammzelltransplantation (LSCT) ist ein aus Japan stammender Ansatz, der weltweit anerkannt ist. Shimazaki et al. berichteten 2002 über die Wirksamkeit der kombinierten Hornhauttransplantation mit LSCT bei GDLD und zeigten, dass sie das rezidivfreie Intervall verlängern kann, indem sie das erneute Einwachsen von Wirtsepithelzellen verhindert 10). Später berichteten auch Movahedan et al. über einen ähnlichen Ansatz 11).
Um die Augenoberfläche mit dem vom Transplantat stammenden Hornhautepithel zu bedecken, wird das Wirtshornhautepithel entfernt und dann eine Limbustransplantation durchgeführt. Postoperativ wird das kontinuierliche Tragen therapeutischer Kontaktlinsen fortgesetzt, um ein Wiederauftreten zu verzögern.
In den letzten Jahren wird auch die Indikation für eine künstliche Hornhaut (Boston Typ I Kpro) geprüft. Da sie nicht über das Wirtshornhautepithel verläuft, kann sie theoretisch eine erneute Amyloidablagerung vermeiden, birgt jedoch Risiken postoperativer Komplikationen wie Infektionen und retroprothetische Membranen.
QTritt die Erkrankung auch nach einer Hornhauttransplantation wieder auf?
A
Bei GDLD ist die Rezidivrate nach Hornhauttransplantation extrem hoch. Es wurde berichtet, dass etwa 97 % innerhalb von 4 Jahren nach einer perforierenden Keratoplastik (PKP) ein Rezidiv erleiden. Die Hauptursache ist die Ersetzung des Transplantatepithels durch Epithelzellen des Empfängers. Als in Japan entwickelte Maßnahmen werden die Kombination einer Limbus-Stammzelltransplantation 10) und das kontinuierliche Tragen therapeutischer Kontaktlinsen 7) eingesetzt, um das Wiederauftreten zu verzögern. Bei der Langzeitbehandlung ist das Ziel, unter der Annahme eines Rezidivs die Sehfunktion zu erhalten und die Operationsintervalle zu verlängern.
6. Pathophysiologie und detaillierter Krankheitsmechanismus
Das für GDLD verantwortliche Gen TACSTD2 ist ein Single-Exon-Gen auf Chromosom 1p32. Im Jahr 1999 identifizierten Tsujikawa et al. es durch Kopplungsanalyse in japanischen Familien als ursächliches Gen 4). Das TACSTD2-Protein spielt eine unverzichtbare Rolle bei der Aufrechterhaltung der Barrierefunktion des Hornhautepithels.
Wenn eine Funktionsverlustmutation im TACSTD2-Gen auftritt, wird die normale intrazelluläre Lokalisation der Tight-Junction-Proteine Claudin 1 und Claudin 7 gestört. Nakatsuka et al. zeigten durch Analyse kultivierter Hornhautepithelzellen und japanischer Familien, dass der Funktionsverlust von TACSTD2 zu einem Verlust der Claudin-Lokalisation an der apikolateralen Junktion und einer verminderten epithelialen Barrierefunktion führt 5). Darüber hinaus berichteten sie 2011 über neue TACSTD2-Mutationen in drei Familien und deren abnormale intrazelluläre Lokalisation 12).
Durch die verminderte epitheliale Barrierefunktion dringen Proteine wie Lactoferrin aus der Tränenflüssigkeit in die Hornhaut ein. Das eingedrungene Lactoferrin bildet Amyloidfibrillen, die sich unter dem Hornhautepithel ablagern. Die Amyloidablagerungen enthalten Lactoferrin, aber diese Krankheit ist keine Anomalie des Lactoferrin-Gens.
Histologisch zeigt die subepitheliale milchige Trübung eine orangerote Färbung mit Kongorot und eine apfelgrüne Doppelbrechung unter dem Polarisationsmikroskop. Im Elektronenmikroskop ist zu beobachten, dass die Tight Junctions des Epithels durch elektronendurchlässige Räume ersetzt sind. Die Ablagerungen dringen auch in die Hornhautlamellen ein und führen zu einer Degeneration von Kollagenfasern und Proteoglykanen.
Für das TACSTD2-Gen wurden über 20 Mutationen berichtet 12). In Japan ist die Q118X-Mutation (Nonsense-Mutation, funktionell null) eine Gründermutation, die über 80 % der pathogenen Chromosomen ausmacht 2). Die Erkrankung tritt typischerweise bei Homozygoten auf, kann aber auch bei compound-heterozygoten Individuen aus Ehen zwischen verschiedenen Familien auftreten. Interessanterweise werden selbst bei derselben Q118X-Homozygotie die vier klinischen Formen (maulbeerartig, bandförmig, kumquatartig und stromale Trübung) gemischt beobachtet 3).
Hornhautamyloidosen werden nach primär oder sekundär, systemisch oder lokal klassifiziert. Die GDLD wird zusammen mit der gittrigen Hornhautdystrophie zu den primären lokalen Amyloidosen gezählt. Die sekundäre lokale Amyloiddegeneration tritt im Zusammenhang mit Trichiasis, Keratokonus, Traumata, langjährigem Kontaktlinsentragen usw. auf und ist Gegenstand der Differentialdiagnose.
Die GDLD ist eine seltene Erkrankung, und es gab das Problem, dass es in einzelnen Einrichtungen an Ärzten mit klinischer Erfahrung mangelte und standardisierte Diagnose- und Behandlungsmethoden fehlten. Die Diagnosekriterien und der Schweregrad wurden von der Forschungsgruppe für Epidemiologie seltener refraktärer Hornhauterkrankungen des Ministeriums für Gesundheit, Arbeit und Soziales und der Forschungsgruppe für die Erstellung und Verbreitung von Behandlungsleitlinien für seltene Erkrankungen des vorderen Augenabschnitts entwickelt 2). Im Jahr 2019 wurde die GDLD als designierte seltene Erkrankung „Gelatinöse tropfenförmige Hornhautdystrophie“ anerkannt, und derzeit werden Minds-konforme (Medical Information Network Distribution Service) Behandlungsleitlinien erstellt 2).
Langfristig stellen Rezidive und mehrere Behandlungen ein Problem dar. Die Kombination von therapeutischen Kontaktlinsen und Limbustransplantation kann das Rezidiv verzögern und das Operationsintervall verlängern, was die Lebensprognose verbessern kann 6, 7, 10).
Berichte über atypische Fälle und einseitige Rezidive
Maeno et al. berichteten über einen klinisch atypischen Fall mit rezidivierender Amyloidablagerung nur in einem Auge und zeigten damit die phänotypische Vielfalt der GDLD13). Der TACSTD2-Gentest spielt bei der Diagnose solcher atypischer Fälle eine entscheidende Rolle 2).
In der Grundlagenforschung wird erwartet, dass die detaillierte Dynamik der Tight-Junction-Moleküle stromabwärts von TACSTD2 aufgeklärt und Therapien entwickelt werden, die auf die Stabilisierung von Claudin abzielen. Klinisch wird die Ausweitung der Indikationen für regenerative medizinische Ansätze wie künstliche Hornhäute, Transplantation von Hornhautepithelblättern und iPS-Zell-abgeleitetes Hornhautepithel untersucht.
Ide T, Nishida K, Maeda N, Tsujikawa M, Yamamoto S, Watanabe H, et al. A spectrum of clinical manifestations of gelatinous drop-like corneal dystrophy in japan. American journal of ophthalmology. 2004;137(6):1081-4. doi:10.1016/j.ajo.2004.01.048. PMID:15183793.
Tsujikawa M, Kurahashi H, Tanaka T, Nishida K, Shimomura Y, Tano Y, et al. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nature genetics. 1999;21(4):420-3. doi:10.1038/7759. PMID:10192395.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Tsujikawa M, et al. Tumor-associated calcium signal transducer 2 is required for the proper subcellular localization of claudin 1 and 7: implications in the pathogenesis of gelatinous drop-like corneal dystrophy. Am J Pathol. 2010;177(3):1344-1355. PMID: 20651236. PMCID: PMC2928967. doi:10.2353/ajpath.2010.100149.
Kawasaki S, Kinoshita S. Clinical and basic aspects of gelatinous drop-like corneal dystrophy. Dev Ophthalmol. 2011;48:97-115. PMID: 21540633. doi:10.1159/000324079.
Maeno S, Soma T, Tsujikawa M, Shigeta R, Kawasaki R, Oie Y, et al. Efficacy of therapeutic soft contact lens in the management of gelatinous drop-like corneal dystrophy. The British journal of ophthalmology. 2020;104(2):241-246. doi:10.1136/bjophthalmol-2018-313809. PMID:31023713.
Hieda O, Kawasaki S, Yamamura K, Nakatsukasa M, Kinoshita S, Sotozono C. Clinical outcomes and time to recurrence of phototherapeutic keratectomy in Japan. Medicine. 2019;98(27):e16216. doi:10.1097/MD.0000000000016216. PMID:31277131; PMCID:PMC6635226.
Shimazaki J, Shimmura S, Tsubota K. Limbal stem cell transplantation for the treatment of subepithelial amyloidosis of the cornea (gelatinous drop-like dystrophy). Cornea. 2002;21(2):177-80. doi:10.1097/00003226-200203000-00010. PMID:11862090.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Nishida K, et al. Two novel mutations of TACSTD2 found in three Japanese gelatinous drop-like corneal dystrophy families with their aberrant subcellular localization. Molecular vision. 2011;17:965-70. PMID:21541270; PMCID:PMC3084224.
Maeno S, Soma T, Nishida K. A Case of Clinically Atypical Gelatinous Drop-like Corneal Dystrophy With Unilateral Recurrent Amyloid Depositions. Cornea. 2022;41(11):1447-1450. doi:10.1097/ICO.0000000000003070. PMID:36219213.
Kopieren Sie den Artikeltext und fügen Sie ihn in den KI-Assistenten Ihrer Wahl ein.
Artikel in die Zwischenablage kopiert
Öffnen Sie unten einen KI-Assistenten und fügen Sie den kopierten Text in den Chat ein.