Желатинозная каплевидная дистрофия роговицы (gelatinous drop-like corneal dystrophy: GDLD) — это наследственное заболевание роговицы, характеризующееся отложением амилоида под эпителием роговицы, приводящее к двустороннему значительному снижению зрения.
Это заболевание впервые было описано в 1914 году Накаидзуми, а в 1932 году Киёсава назвал его «желатинозной каплевидной дегенерацией роговицы», и с тех пор оно известно под этим названием. В классификации IC3D (International Committee for Classification of Corneal Dystrophies) оно классифицируется как эпителиальная дистрофия с аббревиатурой GDLD.
Ген-виновник, TACSTD2 (tumor-associated calcium signal transducer 2), был идентифицирован в 1999 году Цудзикавой и соавт. Это одноэкзонный ген, расположенный на хромосоме 1p324).
Распространенность: В мире редкое заболевание, но в Японии сообщается как относительно частое 1). Считается, что частота возникновения снизилась из-за уменьшения числа близкородственных браков 2)
Региональные различия: Относительно часто встречается в Японии, почти не регистрируется в Европе и Северной Америке
Мутация Q118X: Мутация-основатель у японских пациентов, составляющая более 80% патогенных хромосом 2)
Возраст начала: Часто возникает до 20 лет
В 2019 году заболевание было признано специфическим редким заболеванием «Желатинозная каплевидная дистрофия роговицы» и стало подлежать финансовой поддержке медицинских расходов 2). В рамках исследовательского проекта Министерства здравоохранения, труда и социального обеспечения по рефрактерным заболеваниям были разработаны диагностические критерии и классификация тяжести 2).
QВозникает ли GDLD за пределами Японии?
A
GDLD регистрируется по всему миру, но чаще встречается в Японии. В Европе и Северной Америке случаев почти нет. В гене TACSTD2 зарегистрировано более 20 мутаций, что указывает на генетическую гетерогенность. В Японии мутация Q118X, расположенная на 1p32, часто встречается как мутация-основатель и составляет более 80% патогенных хромосом у японских пациентов.
Часто возникает до 20 лет. Следующие симптомы появляются с детства.
Светобоязнь: Выраженный симптом с самого начала
Ощущение инородного тела: Из-за желатинозных возвышений на поверхности роговицы
Слезотечение: Сопутствует симптомам раздражения
Снижение зрения: Постепенно ухудшается по мере прогрессирования отложения амилоида. Становится заметным после взросления
С возрастом количество и размер амилоидных отложений увеличиваются. Они становятся серовато-белыми или желтыми отложениями и в конечном итоге покрывают большую часть роговицы, особенно в области глазной щели 2). Происходит врастание сосудов с периферии, значительное снижение остроты зрения и боль в глазу, а также добавляются косметические проблемы, что значительно снижает качество жизни пациента.
Клинические признаки (признаки, выявляемые врачом при осмотре)
Помутнение роговицы классифицируется на четыре типа в зависимости от морфологии. Их можно различить при осмотре переднего отрезка с помощью щелевой лампы 2,3).
Тутовый
typical mulberry type: Наиболее типичный тип.
Центральная часть роговицы: Серовато-белые возвышающиеся очаги скапливаются, напоминая тутовую ягоду.
Субэпителиальный амилоид: Молочно-белые полупрозрачные студенистые возвышения увеличиваются от центра к периферии.
Лентовидная кератопатия
band-keratopathy type: Может наблюдаться на ранних стадиях.
Межпальпебральная щель: Поверхностное помутнение. Напоминает лентовидную кератопатию.
Поражение конъюнктивы: Поражения могут также наблюдаться на конъюнктиве.
Кумкватовый
kumquat-like type: Часто встречается в запущенных случаях.
Диффузные желто-белые отложения: Вся роговица становится желтой, напоминая кумкват.
Врастание сосудов: Может сопровождаться поверхностной неоваскуляризацией.
Стромальный помутнение тип
Стромальный помутнение тип : более продвинутая стадия.
Распространение на строму : поражение достигает стромы роговицы.
Ide и соавт. сообщили о подробном клиническом спектре 34 японских случаев и показали, что даже при одной и той же мутации гена TACSTD2 (гомозигота Q118X) сосуществуют четыре фенотипа3).
Кроме того, имеются следующие характерные признаки:
Замедленное окрашивание флуоресцеином (delayed staining) : несмотря на отсутствие повреждения эпителия роговицы, из-за повышенной проницаемости вследствие неполного формирования плотных контактов через несколько минут после инстилляции флуоресцеина наблюдается флуоресценция2)
Истончение эпителия : в области желатинозных возвышений эпителий роговицы истончен
Врастание сосудов : в периферической части роговицы наблюдается поверхностное врастание сосудов
Из-за аномалии гена TACSTD2 теряется нормальная внутриклеточная локализация клаудина-1 и клаудина-7, структурных белков плотных контактов в эпителии роговицы, и снижается барьерная функция эпителия5). В результате такие белки, как лактоферрин из слезной жидкости, проникают в роговицу, образуют амилоидные фибриллы и откладываются под эпителием. Накацука и соавт. с помощью молекулярно-биологического анализа японских семей показали, что TACSTD2 необходим для нормальной локализации клаудинов, и четко установили, что патология GDLD обусловлена дисфункцией плотных контактов5).
Семейный анамнез : из-за аутосомно-рецессивного наследования существует риск развития заболевания, если оба родителя являются носителями.
Японское происхождение: Мутация нонсенс Q118X является основательской мутацией в Японии, составляя более 80% патогенных хромосом2)
Кровнородственный брак: Родители пробанда часто состоят в кровнородственном браке. Однако заболевание может развиться и у компаунд-гетерозигот, рожденных в браках между неродственными семьями.
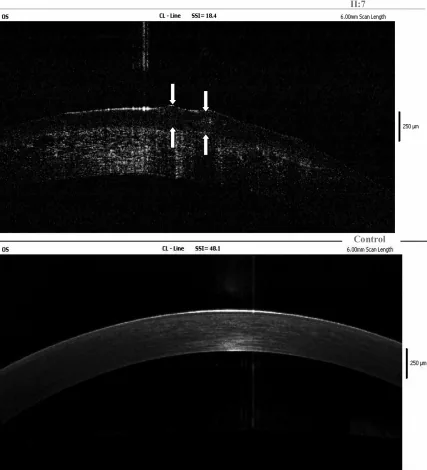
Yang Jing, Chun Liu, Liya Wang A novel TACSTD2 mutation identified in two Chinese brothers with gelatinous drop-like corneal dystrophy 2009 Aug 14 Mol Vis. 2009 Aug 14; 15:1580-1588 Figure 5. PMCID: PMC2728569. License: CC BY.
На ОКТ в частотной области левой роговицы пробанда видны отложения амилоида в эпителии роговицы и поверхностной строме; стрелки указывают на расположение желатинозных каплевидных поражений. Это соответствует отложениям амилоида, рассматриваемым в разделе «4. Диагностика и методы обследования».
Диагностические критерии (исследовательская группа Министерства здравоохранения)
Диагностические критерии GDLD были разработаны исследовательской группой проекта политики в области рефрактерных заболеваний Министерства здравоохранения, труда и социального обеспечения «Создание и распространение клинических рекомендаций по редким заболеваниям переднего сегмента глаза»2). Если по этим критериям установлен диагноз «Definite», заболевание признается редким (орфанным) и подлежит специальному учету.
A. Симптомы (любой из следующих)
Снижение остроты зрения
Светобоязнь
Ощущение инородного тела
Слезотечение
B. Результаты обследования
Наблюдаются серовато-белые возвышающиеся скопления амилоидных отложений (в виде тутовой ягоды) непосредственно под эпителием роговицы в центральной части роговицы и глазной щели обоих глаз.
Наблюдается отсроченное окрашивание (delayed staining): флуоресценция появляется через несколько минут после окрашивания флуоресцеином, несмотря на отсутствие повреждения эпителия роговицы.
Наблюдается поверхностная васкуляризация в периферической области роговицы.
C. Дифференциальный диагноз : исключить вторичный (приобретенный) амилоидоз роговицы и климатическую капельную кератопатию.
D. Внеглазные осложнения : отсутствуют.
E. Генетическое исследование : выявляется аномалия в гене TACSTD2.
Условия для категории Определенный выполняются при наличии одного из следующих пунктов 2):
Случаи, соответствующие критерию D, имеющие один из критериев A, соответствующие критерию B1 и позволяющие исключить заболевания, подлежащие дифференцировке в C.
Случаи, соответствующие критерию D, имеющие один из критериев A, соответствующие критерию B2 или B3, соответствующие критерию E и позволяющие исключить заболевания, подлежащие дифференцировке в C.
B1 (отложения в виде тутовой ягоды) является очень характерным признаком; в типичных случаях диагностика не вызывает затруднений. В нетипичных случаях диагноз ставится на основании комбинации критериев A–C и генетического исследования (E) 2).
Классификация степени тяжести (в соответствии с уведомлением о designated intractable diseases)
Степень тяжести классифицируется по степеням I–IV на основе наилучшей корригированной остроты зрения лучшего глаза 2).
Степень тяжести
Критерий
Финансовая помощь
I степень
Поражение только одного глаза, другой глаз здоров
×
II степень
Поражение обоих глаз, корригированная острота зрения лучшего глаза 0,3 и выше
×
III степень
Поражение обоих глаз, корригированная острота зрения лучшего глаза от 0,1 до 0,3
○
IV степень
Поражение обоих глаз, корригированная острота зрения лучшего глаза менее 0,1
○
При диагнозе Definite заболевание признается редким (из списка designated intractable diseases), и при III степени тяжести и выше можно получить финансовую помощь на медицинские расходы 2). Если из-за вторичной глаукомы и т.п. на лучшем глазу имеется сужение поля зрения (центральное остаточное поле зрения 20 градусов и менее по Goldmann I/4), степень тяжести повышается на одну ступень.
Биомикроскопия с щелевой лампой: Наблюдение серовато-белых возвышающихся поражений от центра роговицы до области глазной щели, различение 4 морфологических типов (тутоподобный, лентовидный, кумкватоподобный, стромальное помутнение).
Тест на проницаемость флуоресцеина (замедленное окрашивание) : из-за дисфункции плотных контактов краситель быстро проникает в ткань роговицы 2)
Оптическая когерентная томография переднего сегмента (AS-OCT) : позволяет неинвазивно оценить глубину и распространение амилоидных отложений в субэпителиальном слое и всей строме роговицы
Генетическое тестирование TACSTD2 : с 2020 финансового года включено в страховое покрытие как генетическое тестирование дистрофий роговицы (D006-20) 2). TACSTD2 является одноэкзонным геном, что облегчает поиск. Особенно полезно для диагностики атипичных случаев
Гистологическое исследование (срез роговицы) : окрашивание конго красным дает оранжево-красный цвет, а под поляризационным микроскопом наблюдается яблочно-зеленое двулучепреломление, подтверждающее амилоид
Вторичный амилоидоз роговицы : отложение амилоида из-за хронического раздражения, такого как трихиаз, энтропион, выпячивание кератоконуса, ношение жестких контактных линз. Отсутствие наследственного анамнеза и наличие фонового хронического воспаления глазной поверхности являются дифференциальными признаками. Может проявляться в виде желатинозных возвышений или решетчатых образований; для подтверждения требуется гистологическое исследование
Климатическая капельная кератопатия : чаще встречается у мужчин старше 40 лет. Наблюдается в пустынных или экстремально холодных регионах, вызвана УФ-излучением и сухостью. Проявляется желтыми или серовато-белыми возвышающимися поражениями роговицы
Ленточная дистрофия роговицы : отложение солей кальция под эпителием. Начинается на периферии в 3 и 9 часов и распространяется к центру
Решетчатая дистрофия роговицы I типа : мутация R124C гена TGFBI, аутосомно-доминантное наследование. Проявляется разветвленными фиброзными помутнениями в строме роговицы
Пятнистая дистрофия роговицы : аномалия гена CHST6, аутосомно-рецессивное наследование. Диффузное помутнение по типу матового стекла
QПокрывается ли генетическое тестирование страховкой?
A
Генетическое тестирование TACSTD2 покрывается страховкой с 2020 года как «Генетическое тестирование дистрофий роговицы (D006-20)». Однако учреждение должно получить сертификацию после создания системы для проведения теста внутри учреждения. Поскольку TACSTD2 является одноэкзонным геном, его легко исследовать, и более 80% японских пациентов имеют мутацию-основатель Q118X, этот тест особенно полезен для диагностики атипичных случаев 2).
Лечение GDLD выбирается в зависимости от степени помутнения и степени нарушения зрения. Из-за наследственного характера заболевания частота рецидивов чрезвычайно высока при любом методе лечения, что является самой большой проблемой 2). Случаи слепоты из-за осложнений множественных пересадок роговицы или вторичной глаукомы нередки.
Искусственные слезы : используются в качестве симптоматической терапии для облегчения симптомов раздражения поверхности.
Непрерывное ношение терапевтических мягких контактных линз (МКЛ) : может подавлять рецидив желатинозных возвышений и увеличивать интервал между операциями.
Непрерывное ношение терапевтических МКЛ рассматривается как консервативное и вспомогательное лечение 6). Maeno и соавт. в 2020 году в проспективном наблюдательном исследовании пациентов с GDLD показали, что ношение терапевтических МКЛ значительно подавляет рецидив серовато-белых до желтых желатинозных возвышений 7). Также рекомендуется для профилактики послеоперационных рецидивов.
Частота рецидивов : После сквозной кератопластики рецидив высок (97% в течение 4 лет). DALK имеет преимущество сохранения эндотелия.
Лимбальная трансплантация
Трансплантация лимбальных стволовых клеток, кератопластика эпителия роговицы : в комбинации с трансплантацией роговицы.
Цель : Покрыть поверхность глаза эпителием роговицы трансплантата и предотвратить повторное врастание эпителия хозяина 10,11).
Из Японии имеется несколько сообщений о долгосрочных результатах ПТК. Ōura и соавт. представили долгосрочные результаты ПТК у пациентов с GDLD, показав полезность в увеличении интервала до рецидива 8). Hieda и соавт. в многоцентровом японском исследовании детально проанализировали время рецидива и клинические исходы после ПТК 9).
Комбинация с трансплантацией лимбальных стволовых клеток (LSCT) является подходом японского происхождения, признанным во всем мире. Shimazaki и соавт. в 2002 году сообщили об эффективности комбинированной трансплантации роговицы с LSCT при GDLD, показав, что она может увеличить интервал до рецидива за счет предотвращения повторного врастания эпителиальных клеток хозяина 10). Позднее Movahedan и соавт. также сообщили о подобном подходе 11).
Для покрытия поверхности глаза эпителием роговицы, полученным от трансплантата, удаляют эпителий роговицы хозяина, а затем проводят лимбальную трансплантацию. После операции продолжают непрерывное ношение лечебных контактных линз, чтобы отсрочить рецидив.
В последние годы также рассматривается применение искусственной роговицы (Boston type I Kpro). Поскольку она не проходит через эпителий роговицы хозяина, теоретически можно избежать повторного отложения амилоида, но существует риск послеоперационных осложнений, таких как инфекции и ретропротезная мембрана.
QВозникает ли рецидив после пересадки роговицы?
A
При GDLD частота рецидивов после пересадки роговицы чрезвычайно высока. Сообщается, что около 97% случаев рецидивируют в течение 4 лет после сквозной кератопластики (PKP). Основной причиной является замещение эпителия трансплантата эпителиальными клетками реципиента. В качестве мер, разработанных в Японии, используются комбинация трансплантации лимбальных стволовых клеток роговицы 10) и непрерывное ношение лечебных контактных линз 7) для отсрочки рецидива. При долгосрочном ведении целью является сохранение зрительной функции и увеличение интервалов между операциями, исходя из того, что рецидив неизбежен.
Ген TACSTD2, вызывающий GDLD, является одноэкзонным геном, расположенным на хромосоме 1p32. В 1999 году Цудзикава и соавторы идентифицировали его как причинный ген с помощью анализа сцепления в японских семьях 4). Белок TACSTD2 играет незаменимую роль в поддержании барьерной функции эпителия роговицы.
Когда в гене TACSTD2 происходит мутация с потерей функции, нарушается нормальная внутриклеточная локализация белков плотных контактов клаудина 1 и клаудина 7. Накацука и соавторы показали на культивируемых эпителиальных клетках роговицы и анализе японских семей, что потеря функции TACSTD2 приводит к потере локализации клаудинов на апиколатеральном стыке и снижению барьерной функции эпителия 5). Кроме того, в 2011 году они сообщили о новых мутациях TACSTD2 в трех семьях и их аномальной внутриклеточной локализации 12).
Из-за снижения барьерной функции эпителия такие белки, как лактоферрин из слезной жидкости, проникают в роговицу. Проникший лактоферрин образует амилоидные фибриллы, которые откладываются под эпителием роговицы. Амилоидные отложения содержат лактоферрин, но это заболевание не является аномалией гена лактоферрина.
Гистологически субэпителиальное молочно-белое помутнение окрашивается в оранжево-красный цвет при окрашивании конго красным и демонстрирует яблочно-зеленое двойное лучепреломление под поляризационным микроскопом. При электронной микроскопии наблюдается, что плотные контакты эпителия заменены электронно-прозрачными пространствами. Отложения также проникают в роговичные пластинки, вызывая дегенерацию коллагеновых волокон и протеогликанов.
Сообщается о более чем 20 мутациях гена TACSTD2 12). В Японии мутация Q118X (нонсенс-мутация, функциональный ноль) является мутацией-основателем, составляющей более 80% патогенных хромосом 2). Заболевание типично развивается у гомозигот, но может также возникать у компаунд-гетерозигот при браках между разными семьями. Интересно, что даже у одних и тех же гомозигот Q118X наблюдаются смешанные четыре клинические формы (тутоподобная, лентовидная, кумкватоподобная и стромальное помутнение) 3).
Амилоидоз роговицы классифицируется как первичный или вторичный, системный или локальный. GDLD вместе с решетчатой дистрофией роговицы относится к первичному локальному амилоидозу. Вторичная локальная амилоидная дегенерация возникает в связи с трихиазом, кератоконусом, травмой, длительным ношением контактных линз и т.д. и является предметом дифференциальной диагностики.
GDLD является редким заболеванием, и существовала проблема нехватки врачей с клиническим опытом в отдельных учреждениях, а также отсутствия стандартных методов диагностики и лечения. Критерии диагностики и классификация степени тяжести были разработаны исследовательской группой по эпидемиологии редких рефрактерных заболеваний роговицы Министерства здравоохранения, труда и социального обеспечения и исследовательской группой по созданию и распространению клинических рекомендаций по редким заболеваниям переднего сегмента 2). В 2019 году GDLD была признана редким заболеванием «Желатинозная каплевидная дистрофия роговицы», и в настоящее время разрабатываются клинические рекомендации, соответствующие Minds (Medical Information Network Distribution Service) 2).
В долгосрочной перспективе рецидивы и многократное лечение становятся проблемой. Комбинация терапевтических контактных линз и лимбальной трансплантации может отсрочить рецидив и увеличить интервал между операциями, что может улучшить пожизненный прогноз 6, 7, 10).
Сообщения об атипичных случаях и одностороннем рецидиве
Maeno и соавт. сообщили о клинически атипичном случае с рецидивирующим отложением амилоида только в одном глазу, демонстрируя фенотипическое разнообразие GDLD 13). Генетическое тестирование гена TACSTD2 играет решающую роль в диагностике таких атипичных случаев 2).
В фундаментальных исследованиях ожидается детальное выяснение динамики молекул плотных контактов ниже TACSTD2 и разработка методов лечения, направленных на стабилизацию клаудина. В клинической практике рассматривается расширение показаний для регенеративных подходов, таких как искусственная роговица, трансплантация листов роговичного эпителия и роговичный эпителий, полученный из iPS-клеток.
Ide T, Nishida K, Maeda N, Tsujikawa M, Yamamoto S, Watanabe H, et al. A spectrum of clinical manifestations of gelatinous drop-like corneal dystrophy in japan. American journal of ophthalmology. 2004;137(6):1081-4. doi:10.1016/j.ajo.2004.01.048. PMID:15183793.
Tsujikawa M, Kurahashi H, Tanaka T, Nishida K, Shimomura Y, Tano Y, et al. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nature genetics. 1999;21(4):420-3. doi:10.1038/7759. PMID:10192395.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Tsujikawa M, et al. Tumor-associated calcium signal transducer 2 is required for the proper subcellular localization of claudin 1 and 7: implications in the pathogenesis of gelatinous drop-like corneal dystrophy. Am J Pathol. 2010;177(3):1344-1355. PMID: 20651236. PMCID: PMC2928967. doi:10.2353/ajpath.2010.100149.
Kawasaki S, Kinoshita S. Clinical and basic aspects of gelatinous drop-like corneal dystrophy. Dev Ophthalmol. 2011;48:97-115. PMID: 21540633. doi:10.1159/000324079.
Maeno S, Soma T, Tsujikawa M, Shigeta R, Kawasaki R, Oie Y, et al. Efficacy of therapeutic soft contact lens in the management of gelatinous drop-like corneal dystrophy. The British journal of ophthalmology. 2020;104(2):241-246. doi:10.1136/bjophthalmol-2018-313809. PMID:31023713.
Hieda O, Kawasaki S, Yamamura K, Nakatsukasa M, Kinoshita S, Sotozono C. Clinical outcomes and time to recurrence of phototherapeutic keratectomy in Japan. Medicine. 2019;98(27):e16216. doi:10.1097/MD.0000000000016216. PMID:31277131; PMCID:PMC6635226.
Shimazaki J, Shimmura S, Tsubota K. Limbal stem cell transplantation for the treatment of subepithelial amyloidosis of the cornea (gelatinous drop-like dystrophy). Cornea. 2002;21(2):177-80. doi:10.1097/00003226-200203000-00010. PMID:11862090.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Nishida K, et al. Two novel mutations of TACSTD2 found in three Japanese gelatinous drop-like corneal dystrophy families with their aberrant subcellular localization. Molecular vision. 2011;17:965-70. PMID:21541270; PMCID:PMC3084224.
Maeno S, Soma T, Nishida K. A Case of Clinically Atypical Gelatinous Drop-like Corneal Dystrophy With Unilateral Recurrent Amyloid Depositions. Cornea. 2022;41(11):1447-1450. doi:10.1097/ICO.0000000000003070. PMID:36219213.
Скопируйте текст статьи и вставьте его в выбранный ИИ-ассистент.
Статья скопирована в буфер обмена
Откройте ИИ-ассистент ниже и вставьте скопированный текст в чат.