La distrofia corneal gelatinosa en gotas (gelatinous drop-like corneal dystrophy: GDLD) es una enfermedad corneal hereditaria en la que se deposita amiloide debajo del epitelio corneal, causando una pérdida visual bilateral significativa.
Esta enfermedad fue reportada por primera vez por Nakazumi en 1914, y desde que Kiyosawa la nombró “degeneración corneal gelatinosa en gotas” en 1932, se ha llamado así. En la clasificación IC3D (Comité Internacional para la Clasificación de Distrofias Corneales), se clasifica como una distrofia epitelial con la abreviatura GDLD.
El gen causante es el gen TACSTD2 (transductor de señal de calcio asociado a tumores 2), identificado por Tsujikawa et al. en 1999, que es un gen de un solo exón ubicado en el cromosoma 1p324).
Prevalencia: Rara a nivel mundial, pero reportada como relativamente común en Japón 1). Se cree que la incidencia está disminuyendo debido a la reducción de matrimonios consanguíneos 2)
Diferencias regionales: Relativamente común en Japón, casi no reportada en Europa y América
Mutación Q118X: Mutación fundadora en pacientes japoneses, que representa más del 80% de los cromosomas causantes de la enfermedad 2)
Edad de inicio: A menudo se desarrolla antes de los 20 años
En 2019 fue designada como enfermedad intratable especificada (distrofia corneal gelatinosa en gotas) y se convirtió en elegible para subsidios de gastos médicos 2). El Proyecto de Investigación de Políticas de Enfermedades Intratables del Ministerio de Salud, Trabajo y Bienestar ha establecido criterios diagnósticos y clasificación de gravedad 2).
Q¿La GDLD también ocurre fuera de Japón?
A
La GDLD se ha reportado en todo el mundo, pero es más común en Japón. Casi no hay casos en Europa y América. Se han reportado más de 20 mutaciones en el gen TACSTD2, lo que indica heterogeneidad genética. En Japón, la mutación Q118X en 1p32 es una mutación fundadora frecuente, que representa más del 80% de los cromosomas causantes de la enfermedad en pacientes japoneses.
A menudo se desarrolla antes de los 20 años. Desde la infancia temprana se presentan los siguientes síntomas.
Fotofobia: Síntoma prominente desde la etapa inicial
Sensación de cuerpo extraño: Debido a elevaciones gelatinosas en la superficie corneal
Lagrimeo: Asociado a síntomas irritativos
Deterioro visual: Empeora gradualmente con la progresión del depósito de amiloide. Se vuelve marcado después de la edad adulta
El número y tamaño de los depósitos de amiloide aumentan con la edad. Se vuelven depósitos de color gris blanquecino a amarillo, y finalmente cubren la mayor parte de la córnea, centrados en la hendidura palpebral 2). Se produce invasión vascular desde la periferia, pérdida visual marcada y dolor ocular, y también surgen problemas estéticos, lo que reduce significativamente la calidad de vida del paciente.
Hallazgos clínicos (hallazgos confirmados por el médico en el examen)
Las opacidades corneales se clasifican en cuatro tipos según su morfología. Estos se pueden distinguir mediante la observación del segmento anterior con un microscopio de lámpara de hendidura2,3).
Tipo mora
tipo mora típico (typical mulberry type): El tipo más típico.
Córnea central: Lesiones elevadas de color gris blanquecino se agrupan, asemejándose a la apariencia de una mora.
Amiloide subepitelial: Elevaciones gelatinosas de color blanco lechoso y semitransparentes aumentan desde el centro hacia la periferia.
Tipo queratopatía en banda
tipo queratopatía en banda (band-keratopathy type): Puede observarse en etapas tempranas.
Zona interpalpebral: Se observan opacidades superficiales. Los hallazgos se asemejan a la queratopatía en banda.
Lesiones conjuntivales: También pueden presentarse lesiones en la conjuntiva.
Tipo kumquat
tipo kumquat (kumquat-like type): Común en casos avanzados.
Depósitos difusos de color blanco amarillento: Toda la córnea se vuelve amarilla, asemejándose a un kumquat.
Invasión vascular: Puede acompañarse de neovascularización superficial.
Tipo de opacidad del estroma
tipo de opacidad del estroma: etapa más avanzada.
Extensión al estroma: la lesión afecta al estroma corneal.
Invasión vascular: las elevaciones gelatinosas de color blanco lechoso a amarillo se acompañan de invasión vascular.
Ide y colaboradores reportaron el espectro clínico detallado de 34 casos en Japón y demostraron que incluso con la misma mutación del gen TACSTD2 (homocigoto Q118X) coexisten cuatro fenotipos3).
Además, se observan los siguientes hallazgos característicos:
Tinción retardada con fluoresceína (delayed staining): a pesar de la ausencia de daño epitelial corneal, se observa fluorescencia minutos después de la instilación de fluoresceína debido al aumento de la permeabilidad por la formación deficiente de uniones estrechas2)
Adelgazamiento epitelial: el epitelio corneal está adelgazado en las áreas de elevaciones gelatinosas
Invasión vascular: se observa invasión vascular superficial en la periferia corneal
La anomalía del gen TACSTD2 provoca la pérdida de la localización intracelular normal de Claudina 1 y Claudina 7, proteínas constituyentes de las uniones estrechas en el epitelio corneal, lo que reduce la función de barrera epitelial5). Como resultado, proteínas como la lactoferrina de la lágrima penetran en la córnea, forman fibrillas amiloides y se depositan bajo el epitelio. Nakatsukasa y colaboradores demostraron molecularmente, mediante el análisis de familias japonesas, que TACSTD2 es esencial para la localización normal de las claudinas, y aclararon que la patología de la GDLD se debe a una disfunción de las uniones estrechas5).
Antecedentes familiares: al ser una herencia autosómica recesiva, existe riesgo de desarrollar la enfermedad si ambos padres son portadores.
Ascendencia japonesa: La mutación sin sentido Q118X representa más del 80% de los cromosomas patogénicos como mutación fundadora en Japón 2)
Matrimonio consanguíneo: Por lo general, los padres del probando son consanguíneos. Sin embargo, también puede ocurrir en heterocigotos compuestos de matrimonios entre diferentes familias.
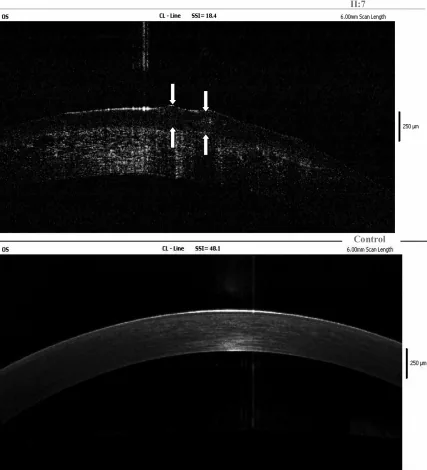
Yang Jing, Chun Liu, Liya Wang A novel TACSTD2 mutation identified in two Chinese brothers with gelatinous drop-like corneal dystrophy 2009 Aug 14 Mol Vis. 2009 Aug 14; 15:1580-1588 Figure 5. PMCID: PMC2728569. License: CC BY.
Los hallazgos de OCT de dominio de Fourier de la córnea izquierda del probando muestran depósitos de amiloide en el epitelio corneal y el estroma superficial, y las flechas indican la ubicación de las lesiones gelatinosas en gotas. Esto corresponde a los depósitos de amiloide discutidos en la sección “4. Diagnóstico y métodos de prueba.”
Criterios diagnósticos (grupo de investigación del Ministerio de Salud, Trabajo y Bienestar)
Los criterios diagnósticos para GDLD han sido establecidos por el grupo de investigación del Ministerio de Salud, Trabajo y Bienestar “Investigación para la creación y difusión de guías de práctica clínica para enfermedades intratables del segmento anterior” 2). Si un paciente es clasificado como Definitivo según estos criterios, se convierte en sujeto de enfermedades intratables designadas.
A. Síntomas (cualquiera de los siguientes)
Disminución de la agudeza visual
Fotofobia
Sensación de cuerpo extraño
Lagrimeo
B. Hallazgos de laboratorio
Se observan depósitos amiloides elevados de color blanco grisáceo (en forma de mora) debajo del epitelio corneal en la córnea central hasta la hendidura palpebral de ambos ojos.
Se observa tinción retardada (delayed staining) unos minutos después de la tinción con fluoresceína, a pesar de la ausencia de daño epitelial corneal.
Se observa invasión vascular superficial en la córnea periférica.
C. Diagnóstico diferencial: Excluir amiloidosis corneal secundaria (adquirida) y queratopatía climática en gotas (climatic droplet keratopathy).
D. Complicaciones extraoculares: Ninguna.
E. Pruebas genéticas: Se encuentran anomalías en el gen TACSTD2.
Definitivo se cumple si se satisface alguna de las siguientes condiciones2).
Casos que cumplen D, presentan cualquiera de A, presentan B1, y pueden excluir las enfermedades a diferenciar en C.
Casos que cumplen D, presentan cualquiera de A, presentan B2 o B3, presentan E, y pueden excluir las enfermedades a diferenciar en C.
B1 (depósitos en forma de mora) es un hallazgo muy característico, y el diagnóstico no es difícil en casos típicos. En casos atípicos, el diagnóstico se realiza combinando A a C con pruebas genéticas (E)2).
Clasificación de gravedad (para la notificación de enfermedades intratables designadas)
La gravedad se clasifica en grados I a IV según la mejor agudeza visual corregida en el ojo con mejor visión2).
Gravedad
Criterio
Subsidio de gastos médicos
Grado I
Solo un ojo afectado, el otro ojo sano
×
Grado II
Ambos ojos afectados, mejor agudeza visual corregida en el ojo mejor ≥ 0.3
×
Grado III
Ambos ojos afectados, mejor agudeza visual corregida en el ojo mejor ≥ 0.1 y < 0.3
○
Grado IV
Ambos ojos afectados, mejor agudeza visual corregida en el ojo mejor < 0.1
○
Un diagnóstico de Definite califica como enfermedad intratable designada, y se pueden recibir subsidios de gastos médicos para el grado de severidad III o superior 2). Si el ojo mejor presenta estrechamiento del campo visual (campo visual central residual ≤ 20 grados con el estímulo Goldmann I/4) debido a glaucoma secundario, etc., la severidad se incrementa en un nivel.
Microscopía con lámpara de hendidura: Observe lesiones elevadas de color gris-blanco desde la córnea central hasta el área de la fisura palpebral, y diferencie cuatro tipos (tipo mora, tipo banda, tipo kumquat y tipo opacidad parenquimatosa).
Prueba de penetración de fluoresceína (tinción retardada): Debido a la disfunción de las uniones estrechas, el colorante penetra rápidamente en el tejido corneal 2)
Prueba genética TACSTD2: Cubierta por el seguro desde el año fiscal 2020 como prueba genética de distrofia corneal (D006-20) 2). TACSTD2 es un gen de un solo exón, lo que facilita su análisis. Particularmente útil para diagnosticar casos atípicos
Examen histológico (sección corneal): El amiloide se confirma mediante tinción rojo anaranjado con rojo Congo y birrefringencia verde manzana bajo microscopio de luz polarizada
Amiloidosis corneal secundaria: Depósitos de amiloide debidos a irritación crónica como triquiasis, entropión, protrusión del queratocono o uso de lentes de contacto rígidas. Los rasgos distintivos incluyen ausencia de antecedentes familiares y antecedentes de inflamación crónica de la superficie ocular. Puede presentar hallazgos similares a gotas gelatinosas o enrejado; se necesita examen histológico para confirmación
Queratopatía climática por gotitas: Más común en hombres mayores de 40 años. Se encuentra en regiones desérticas o extremadamente frías, causada por radiación ultravioleta y sequedad. Se presenta con lesiones corneales elevadas de color amarillo a gris blanquecino
Degeneración corneal en banda: Depósitos de sales de calcio en el subepitelio. Comienza en la periferia a las 3 y 9 en punto y progresa centralmente
Distrofia corneal reticular tipo I: Herencia autosómica dominante debida a mutación R124C del gen TGFBI. Se presenta con opacidades filamentosas ramificadas en el estroma corneal
Distrofia corneal macular: Anomalía del gen CHST6, herencia autosómica recesiva. Opacidades difusas en vidrio esmerilado
Q¿La prueba genética está cubierta por el seguro?
A
La prueba genética TACSTD2 está cubierta por el seguro desde 2020 como “Prueba genética de distrofia corneal (D006-20)”. Sin embargo, el centro debe estar certificado y tener la capacidad de realizar la prueba. TACSTD2 es un gen de un solo exón, lo que facilita su análisis, y más del 80% de los pacientes japoneses tienen la mutación fundadora Q118X, lo que la hace particularmente útil para diagnosticar casos atípicos 2).
El tratamiento de la GDLD se selecciona según la extensión de las opacidades y el grado de deterioro visual. Al ser una enfermedad hereditaria, el principal desafío es la tasa de recurrencia extremadamente alta con cualquier tratamiento 2). No son pocos los casos que llevan a la ceguera debido a complicaciones de múltiples trasplantes de córnea o glaucoma secundario.
Lágrimas artificiales: Se utilizan como tratamiento sintomático para aliviar la irritación superficial.
Uso continuo de lentes de contacto blandas terapéuticas (SCL): Puede suprimir la recurrencia de lesiones elevadas gelatinosas y prolongar el intervalo entre cirugías.
El uso continuo de SCL terapéuticas se considera un tratamiento conservador y coadyuvante 6). En 2020, Maeno et al. demostraron en un estudio observacional prospectivo en pacientes con GDLD que el uso de SCL terapéuticas suprimió significativamente la recurrencia de lesiones elevadas gelatinosas de color blanco grisáceo a amarillo 7). También se recomienda para la prevención de recurrencias postoperatorias.
Queratectomía terapéutica con láser excímero: Primera opción para opacidades superficiales.
Indicaciones: Elevaciones gelatinosas superficiales de leves a moderadas. Combinado con raspado manual. Resultados a largo plazo 8,9).
Trasplante de Córnea
Superficial, lamelar anterior profundo (DALK) y penetrante (PKP): Indicado en casos avanzados.
Tasa de recurrencia: La recurrencia después del trasplante penetrante es alta, del 97% en 4 años. DALK tiene la ventaja de preservar el endotelio.
Trasplante de Limbo Corneal
Trasplante de células madre limbares / queratoplastia epitelial: Combinado con trasplante de córnea.
Objetivo: Cubrir la superficie ocular con epitelio corneal derivado del injerto y prevenir la reinvasión del epitelio del huésped 10,11).
Se han reportado múltiples resultados a largo plazo de PTK desde Japón. Oura et al. reportaron resultados a largo plazo de PTK para casos de GDLD, mostrando su utilidad para prolongar el intervalo libre de recurrencia 8). Hieda et al. analizaron detalladamente el momento de la recurrencia y los resultados clínicos después de PTK en un estudio multicéntrico japonés 9).
La combinación con trasplante de células madre limbares (LSCT) es un enfoque de origen japonés reconocido internacionalmente. Shimazaki et al. reportaron en 2002 la eficacia del trasplante de córnea combinado con LSCT para GDLD, mostrando que puede prolongar el intervalo libre de recurrencia al suprimir la reinvasión de las células epiteliales del huésped 10). Posteriormente, Movahedan et al. también reportaron un enfoque similar 11).
Para cubrir la superficie ocular con epitelio corneal derivado del injerto, se elimina el epitelio corneal del huésped y luego se realiza un trasplante de limbo. Después de la cirugía, se continúa con el uso continuo de LCT terapéuticas para retrasar aún más la recurrencia.
En los últimos años, también se ha considerado la indicación de córnea artificial (Kpro tipo I de Boston). Dado que no involucra el epitelio corneal del huésped, teóricamente se puede evitar la redeposición de amiloide, pero existen riesgos de complicaciones postoperatorias como infección y membrana retroprotésica.
Q¿El trasplante de córnea también provoca recurrencia?
A
En la GDLD, la tasa de recurrencia después del trasplante de córnea es extremadamente alta. Se ha informado que aproximadamente el 97% recurre dentro de los 4 años posteriores a la queratoplastia penetrante (PKP). La causa principal es el reemplazo del epitelio del injerto por células epiteliales del receptor. Como medida de origen japonés, se utiliza la combinación de trasplante de células madre del limbo corneal 10) y el uso continuo de LCT terapéuticas 7) para retrasar la recurrencia. En el manejo a largo plazo, el objetivo es mantener la función visual y prolongar el intervalo entre cirugías, asumiendo la recurrencia.
TACSTD2, el gen causante de la GDLD, es un gen de un solo exón ubicado en el cromosoma 1p32. En 1999, Tsujikawa et al. lo identificaron como el gen causante mediante análisis de ligamiento de familias japonesas 4). La proteína TACSTD2 desempeña un papel esencial en el mantenimiento de la función de barrera del epitelio corneal.
Cuando ocurre una mutación de pérdida de función en el gen TACSTD2, se altera la localización intracelular normal de Claudina 1 y Claudina 7, que son proteínas constituyentes de las uniones estrechas. Nakatsukasa et al. demostraron mediante análisis de células epiteliales corneales cultivadas y familias japonesas que la pérdida de función de TACSTD2 conduce a la pérdida de localización de Claudina en la unión apicolateral y a una reducción de la función de barrera epitelial 5). Además, en 2011, informaron nuevas mutaciones de TACSTD2 en tres familias y su localización intracelular anormal 12).
Debido a la reducción de la función de barrera epitelial, proteínas como la lactoferrina en las lágrimas invaden la córnea. La lactoferrina invasora forma fibrillas de amiloide y se deposita debajo del epitelio corneal. Aunque los depósitos de amiloide contienen lactoferrina, esta enfermedad no se debe a una anomalía en el gen de la lactoferrina.
Histológicamente, las opacidades subepiteliales de color blanco lechoso se tiñen de naranja-rojo con rojo Congo y muestran birrefringencia verde manzana bajo microscopía de luz polarizada. La microscopía electrónica revela que las uniones estrechas del epitelio son reemplazadas por espacios electrónicamente translúcidos. Los depósitos también invaden las láminas corneales, causando degeneración de las fibras de colágeno y proteoglicanos.
Se han reportado más de 20 mutaciones en el gen TACSTD2 12). En Japón, la mutación Q118X (mutación sin sentido, nula funcional) representa más del 80% de los cromosomas patogénicos como mutación fundadora 2). Típicamente se presenta en homocigotos, pero también puede ocurrir en heterocigotos compuestos por matrimonios entre diferentes familias. Curiosamente, incluso entre individuos con la misma homocigosidad Q118X, se observan cuatro subtipos clínicos (tipo mora, en banda, tipo kumquat y tipo opacidad estromal) 3).
La amiloidosis corneal se clasifica como primaria o secundaria, y sistémica o localizada. La GDLD, junto con la distrofia corneal reticular, se clasifica como una amiloidosis localizada primaria. La degeneración amiloide localizada secundaria ocurre con triquiasis, queratocono, traumatismo, uso prolongado de lentes de contacto, etc., y es un diagnóstico diferencial.
La GDLD es una enfermedad rara, y existía el desafío de que pocos médicos en instituciones individuales tenían experiencia clínica, y no se habían establecido métodos estándar de diagnóstico y tratamiento. El Proyecto de Investigación de Políticas de Enfermedades Intratables del Ministerio de Salud, Trabajo y Bienestar, “Grupo de Investigación de Encuesta Epidemiológica de Enfermedades Corneales Raras e Intratables” y el “Grupo de Investigación para el Desarrollo y Difusión de Guías de Práctica Clínica para Enfermedades Intratables del Segmento Anterior” formularon criterios diagnósticos y clasificación de gravedad 2). En 2019, fue designada como enfermedad intratable especificada “Distrofia Corneal en Gotas Gelatinosas,” y actualmente se está desarrollando una guía de práctica clínica conforme a Minds (Servicio de Distribución de Red de Información Médica) 2).
A largo plazo, la recurrencia y los múltiples tratamientos son problemáticos. La combinación de LCT terapéutico y trasplante de limbo puede retrasar la recurrencia y prolongar el intervalo entre cirugías, mejorando potencialmente el pronóstico a largo plazo 6, 7, 10).
Reportes de Casos Atípicos y Recurrencia Unilateral
Maeno et al. reportaron un caso clínicamente atípico que mostraba depósito amiloide recurrente en un solo ojo, demostrando la diversidad fenotípica de la GDLD 13). Las pruebas genéticas de TACSTD2 juegan un papel decisivo en el diagnóstico de estos casos atípicos 2).
En la investigación básica, se espera la elucidación detallada de la dinámica de las moléculas de unión estrecha aguas abajo de TACSTD2 y el desarrollo de terapias dirigidas a la estabilización de Claudina. En el ámbito clínico, se está considerando la expansión de las indicaciones para enfoques de medicina regenerativa como córneas artificiales, trasplante de láminas de epitelio corneal y epitelio corneal derivado de células iPS.
Ide T, Nishida K, Maeda N, Tsujikawa M, Yamamoto S, Watanabe H, et al. A spectrum of clinical manifestations of gelatinous drop-like corneal dystrophy in japan. American journal of ophthalmology. 2004;137(6):1081-4. doi:10.1016/j.ajo.2004.01.048. PMID:15183793.
Tsujikawa M, Kurahashi H, Tanaka T, Nishida K, Shimomura Y, Tano Y, et al. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nature genetics. 1999;21(4):420-3. doi:10.1038/7759. PMID:10192395.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Tsujikawa M, et al. Tumor-associated calcium signal transducer 2 is required for the proper subcellular localization of claudin 1 and 7: implications in the pathogenesis of gelatinous drop-like corneal dystrophy. Am J Pathol. 2010;177(3):1344-1355. PMID: 20651236. PMCID: PMC2928967. doi:10.2353/ajpath.2010.100149.
Kawasaki S, Kinoshita S. Clinical and basic aspects of gelatinous drop-like corneal dystrophy. Dev Ophthalmol. 2011;48:97-115. PMID: 21540633. doi:10.1159/000324079.
Maeno S, Soma T, Tsujikawa M, Shigeta R, Kawasaki R, Oie Y, et al. Efficacy of therapeutic soft contact lens in the management of gelatinous drop-like corneal dystrophy. The British journal of ophthalmology. 2020;104(2):241-246. doi:10.1136/bjophthalmol-2018-313809. PMID:31023713.
Hieda O, Kawasaki S, Yamamura K, Nakatsukasa M, Kinoshita S, Sotozono C. Clinical outcomes and time to recurrence of phototherapeutic keratectomy in Japan. Medicine. 2019;98(27):e16216. doi:10.1097/MD.0000000000016216. PMID:31277131; PMCID:PMC6635226.
Shimazaki J, Shimmura S, Tsubota K. Limbal stem cell transplantation for the treatment of subepithelial amyloidosis of the cornea (gelatinous drop-like dystrophy). Cornea. 2002;21(2):177-80. doi:10.1097/00003226-200203000-00010. PMID:11862090.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Nishida K, et al. Two novel mutations of TACSTD2 found in three Japanese gelatinous drop-like corneal dystrophy families with their aberrant subcellular localization. Molecular vision. 2011;17:965-70. PMID:21541270; PMCID:PMC3084224.
Maeno S, Soma T, Nishida K. A Case of Clinically Atypical Gelatinous Drop-like Corneal Dystrophy With Unilateral Recurrent Amyloid Depositions. Cornea. 2022;41(11):1447-1450. doi:10.1097/ICO.0000000000003070. PMID:36219213.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.