Dut benzeri
Tipik dut tipi: En yaygın hastalık tipi.
Kornea merkezi: Grimsi beyaz kabarık lezyonlar kümeleşerek dut görünümüne benzer.
Subepitelyal amiloid: Süt beyazı, yarı saydam jelatinimsi kabarıklıklar merkezden çevreye doğru artar.

Jelatinöz damla benzeri kornea distrofisi (Gelatinous drop-like corneal dystrophy: GDLD), kornea epiteli altında amiloid birikmesi sonucu iki taraflı belirgin görme azalmasına yol açan kalıtsal bir kornea hastalığıdır.
Bu hastalık ilk kez 1914’te Nakazumi tarafından bildirilmiş ve 1932’de Kiyozawa tarafından “jelatinöz damla benzeri kornea dejenerasyonu” olarak adlandırılmıştır. IC3D (Uluslararası Kornea Distrofileri Sınıflandırma Komitesi) sınıflamasında epitelyal distrofiler grubunda yer alır ve kısaltması GDLD’dir.
Sorumlu gen, 1999’da Tsujikawa ve arkadaşları tarafından tanımlanan TACSTD2 (tümörle ilişkili kalsiyum sinyal dönüştürücü 2) genidir ve kromozom 1p32’de yer alan tek ekzonlu bir gendir4).
2019 yılında nadir hastalık olarak tanınmış ve tıbbi yardım kapsamına alınmıştır 2). Sağlık, Çalışma ve Refah Bakanlığı’nın dirençli hastalık politika araştırma projesi tarafından tanı kriterleri ve şiddet sınıflandırması oluşturulmuştur 2).
GDLD dünya çapında bildirilmiştir, ancak Japonya’da nispeten sık görülür. Avrupa ve Amerika’da neredeyse hiç vaka yoktur. TACSTD2 geninde 20’den fazla mutasyon rapor edilmiştir ve genetik heterojenlik gösterir. Japonya’da, 1p32’de bulunan Q118X mutasyonu yüksek sıklıkta bir kurucu mutasyon olarak görülür ve Japon hastaların hastalık kromozomlarının %80’inden fazlasını oluşturduğu düşünülmektedir.
Genellikle 20 yaşına kadar ortaya çıkar. Çocukluktan itibaren aşağıdaki belirtiler görülür.
Yaşla birlikte amiloid birikintilerinin sayısı ve boyutu artar. Grimsi beyazdan sarıya dönen birikintiler, sonunda göz kapağı aralığı merkezli olarak korneanın büyük kısmını kaplar 2). Periferden damar invazyonu, belirgin görme azalması ve göz ağrısı oluşur; kozmetik sorunlar da eklenince hastanın yaşam kalitesi önemli ölçüde düşer.
Kornea opasitesinin şekline göre dört tip hastalık sınıflandırılır. Bunlar yarık lamba mikroskobu ile ön segment muayenesinde ayırt edilebilir 2,3).
Dut benzeri
Tipik dut tipi: En yaygın hastalık tipi.
Kornea merkezi: Grimsi beyaz kabarık lezyonlar kümeleşerek dut görünümüne benzer.
Subepitelyal amiloid: Süt beyazı, yarı saydam jelatinimsi kabarıklıklar merkezden çevreye doğru artar.
Bant keratopati tipi
Bant keratopati tipi: Erken evrelerde görülebilir.
Göz kapağı aralığı: Yüzeyel tabakalarda opasite izlenir. Bant keratopatiye benzer bulgular gösterir.
Konjonktiva lezyonları: Konjonktivada da lezyonlar görülebilir.
Kumkat benzeri
Kumkat benzeri tip: İleri vakalarda sık görülür.
Yaygın sarı-beyaz birikintiler: Tüm kornea sarararak kumkat görünümü alır.
Damar invazyonu: Yüzeyel neovaskülarizasyon eşlik edebilir.
Stromal opasite tipi
Stromal opasite tipi: Daha ileri evre.
Stromaya yayılım: Lezyon kornea stromasına uzanır.
Damar invazyonu: Süt beyazı ila sarı renkli jelatinimsi kabarıklıklara damar invazyonu eşlik eder.
Ide ve arkadaşları Japonya’daki 34 olgunun ayrıntılı klinik spektrumunu bildirmiş ve aynı TACSTD2 gen mutasyonunda (Q118X homozigot) bile dört fenotipin bir arada bulunabileceğini göstermiştir3).
Ayrıca aşağıdaki karakteristik bulgular vardır:
GDLD, TACSTD2 genindeki fonksiyon kaybı mutasyonları sonucu ortaya çıkar4).
TACSTD2 gen anormalliği nedeniyle, kornea epitelinde sıkı bağlantıların yapısal proteinleri olan Claudin 1 ve Claudin 7’nin normal hücre içi lokalizasyonu kaybolur ve epitel bariyer fonksiyonu azalır5). Sonuçta gözyaşındaki laktoferrin gibi proteinler korneaya girerek amiloid fibrilleri oluşturur ve epitel altında birikir. Nakatsuka ve arkadaşları Japon ailelerin analiziyle TACSTD2’nin normal claudin lokalizasyonu için gerekli olduğunu moleküler biyolojik olarak göstermiş ve GDLD patofizyolojisinin sıkı bağlantı fonksiyon bozukluğundan kaynaklandığını netleştirmiştir5).
GDLD için tanı kriterleri, Sağlık, Çalışma ve Refah Bakanlığı’nın «Ön segment nadir hastalıkları için klinik kılavuz oluşturma ve yaygınlaştırma araştırması» proje grubu tarafından oluşturulmuştur2). Bu tanı kriterlerine göre Kesin (Definite) olarak değerlendirilirse, belirlenmiş nadir hastalık kapsamına girer.
A. Belirtiler (aşağıdakilerden herhangi biri)
B. Laboratuvar bulguları
C. Ayırıcı tanı: Sekonder korneal amiloidoz ve klimatik droplet keratopati dışlanmalıdır.
D. Oküler dışı komplikasyonlar: Yok.
E. Genetik test: TACSTD2 geninde anormallik saptanır.
Kesin (Definite) tanı için aşağıdaki koşullardan birinin karşılanması gerekir2).
B1 (dut benzeri birikimler) oldukça karakteristik bir bulgudur ve tipik olgularda tanı zor değildir. Atipik olgularda tanı, A-C ve genetik test (E) kombinasyonu ile konur2).
Şiddet, daha iyi olan gözün düzeltilmiş görme keskinliğine göre I-IV derecelerine ayrılır2).
| Şiddet | Kriter | Tıbbi mali yardım |
|---|---|---|
| Derece I | Sadece bir göz etkilenmiş, diğer göz sağlıklı | × |
| Derece II | Her iki göz etkilenmiş, iyi olan gözde düzeltilmiş görme keskinliği 0.3 veya daha iyi | × |
| Derece III | Her iki göz etkilenmiş, iyi olan gözde düzeltilmiş görme keskinliği 0.1 veya daha iyi ancak 0.3’ten az | ○ |
| Derece IV | Her iki göz etkilenmiş, iyi olan gözde düzeltilmiş görme keskinliği 0.1’den az | ○ |
Definite tanısı konulduğunda belirlenmiş nadir hastalık kapsamına girer ve şiddet derecesi III veya üzeri olduğunda tıbbi masraf desteği alınabilir2). Sekonder glokom vb. nedeniyle iyi olan gözde görme alanı daralması (Goldmann I/4 hedefi ile merkezi rezidüel görme alanı 20 derece veya daha az) varsa, şiddet bir derece artırılır.
TACSTD2 gen testi 2020 yılından itibaren “Kornea Distrofisi Gen Testi (D006-20)” olarak sigorta kapsamına alınmıştır. Ancak, testi yapabilecek altyapıya sahip olmak ve tesis onayı almak gereklidir. TACSTD2 tek ekzonlu bir gen olduğu için taranması kolaydır ve Japon hastaların %80’inden fazlası Q118X kurucu mutasyonuna sahip olduğundan, atipik vakaların tanısında özellikle faydalıdır2).
GDLD tedavisi, opasitenin yaygınlığına ve görme bozukluğunun derecesine göre seçilir. Kalıtsal bir hastalık olduğu için, herhangi bir tedavi yönteminde nüks oranının son derece yüksek olması en büyük sorundur2). Birden fazla kornea nakline bağlı komplikasyonlar ve sekonder glokom nedeniyle körlüğe yol açan vakalar az değildir.
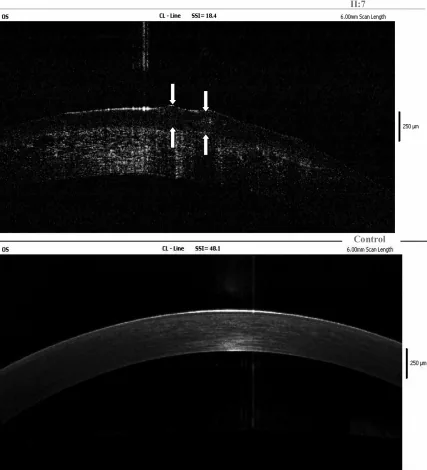
Terapötik SCL’lerin sürekli kullanımı konservatif ve yardımcı bir tedavi olarak değerlendirilir 6). Maeno ve ark. 2020’de GDLD hastalarında yaptıkları prospektif gözlemsel çalışmada, terapötik SCL kullanımının grimsi-beyaz ila sarı jelatinimsi kabarık lezyonların nüksünü anlamlı şekilde baskıladığını gösterdi 7). Postoperatif nüks önlemede de önerilir.
PTK
Eksimer lazer terapötik yüzeyel keratektomi: Yüzeyel opasitelerde ilk seçenek.
Endikasyon: Hafif ila orta dereceli yüzeyel jelatinimsi kabarıklıklar. Manuel küretaj ile kombine edilir. Uzun dönem sonuçlar 8,9).
Kornea Nakli
Limbus Kornea Nakli
Limbus kök hücre nakli ve epitelyal keratoplasti: Kornea nakline ek olarak kullanılır.
Amaç: Greft kaynaklı kornea epiteli ile oküler yüzeyi kaplamak ve konak epitelinin yeniden invazyonunu önlemek 10,11).
PTK’nın uzun dönem sonuçları Japonya’dan çeşitli raporlarda bildirilmiştir. Ōura ve ark. GDLD vakalarında PTK’nın uzun dönem sonuçlarını göstermiş ve nükse kadar geçen süreyi uzatmada faydalı olduğunu rapor etmiştir 8). Hieda ve ark. Japonya’da çok merkezli bir çalışmada PTK sonrası nüks zamanı ve klinik sonuçları detaylı olarak analiz etmiştir 9).
Limbus kök hücre nakli (LSCT) kombinasyonu, Japonya kaynaklı bir yaklaşım olarak dünya çapında değerlendirilmektedir. Shimazaki ve ark. 2002’de GDLD için LSCT ile kombine kornea naklinin etkinliğini rapor etmiş ve konak epitel hücrelerinin yeniden invazyonunu baskılayarak nükse kadar geçen süreyi uzatabildiğini göstermiştir 10). Daha sonra Movahedan ve ark. da benzer bir yaklaşım rapor etmiştir 11).
Göz yüzeyini greft kaynaklı kornea epiteli ile kaplamak için, önce konak kornea epiteli çıkarılır ve ardından limbal transplantasyon yapılır. Ameliyat sonrası tedavi amaçlı kontakt lensin sürekli kullanımına devam edilir ve nüks geciktirilir.
Son yıllarda yapay kornea (Boston tip I Kpro) uygulaması da değerlendirilmektedir. Konak kornea epitelini gerektirmediği için teorik olarak amiloid birikimini önler, ancak enfeksiyon, yapay kornea arka membranı gibi postoperatif komplikasyon riskleri vardır.
GDLD’de kornea nakli sonrası nüks oranı oldukça yüksektir. Tam kat kornea nakli (PKP) sonrası 4 yıl içinde yaklaşık %97’sinin nüks ettiği bildirilmiştir. Ana neden, alıcının epitel hücrelerinin greft epiteli ile yer değiştirmesidir. Japonya kaynaklı bir önlem olarak, kornea limbal kök hücre naklinin birlikte kullanımı 10) ve tedavi amaçlı kontakt lensin sürekli kullanımı 7) ile nüks geciktirilmeye çalışılmaktadır. Uzun dönem yönetimde, nüksün kaçınılmaz olduğu kabul edilerek görme fonksiyonunun korunması ve cerrahi aralıkların uzatılması hedeflenir.
GDLD’nin sorumlu geni olan TACSTD2, kromozom 1p32’de yer alan tek ekzonlu bir gendir. 1999 yılında Tsujikawa ve arkadaşları, Japon ailelerinde yaptıkları bağlantı analizi ile bu geni sorumlu gen olarak tanımlamıştır 4). TACSTD2 proteini, kornea epitelinin bariyer fonksiyonunun sürdürülmesinde vazgeçilmez bir rol oynar.
TACSTD2 geninde fonksiyon kaybına yol açan mutasyonlar, sıkı bağlantıların yapısal proteinleri olan Claudin 1 ve Claudin 7’nin normal hücre içi lokalizasyonunu bozar. Nakatsuka ve arkadaşları, kültüre edilmiş kornea epitel hücreleri ve Japon ailelerinin analizi ile TACSTD2 fonksiyon kaybının Claudin’lerin apikolateral bağlantı bölgesindeki lokalizasyonunu kaybettirdiğini ve epitel bariyer fonksiyonunu azalttığını göstermiştir 5). Ayrıca 2011 yılında üç ailede yeni TACSTD2 mutasyonları ve anormal hücre içi lokalizasyonlarını rapor etmişlerdir 12).
Epitel bariyer fonksiyonunun azalması, gözyaşındaki laktoferrin gibi proteinlerin korneaya girmesine neden olur. Giren laktoferrin, amiloid fibrilleri oluşturarak kornea epiteli altında birikir. Amiloid birikintileri laktoferrin içerir, ancak bu hastalık laktoferrin genindeki bir anormallikten kaynaklanmaz.
Histolojik olarak, subepitelyal süt beyazı opasite Kongo kırmızısı boyamasıyla turuncu-kırmızı renk alır ve polarizasyon mikroskobunda elma yeşili çift kırılım gösterir. Elektron mikroskobunda, epitelyal sıkı bağlantıların elektron saydam boşluklarla yer değiştirdiği gözlenir. Birikintiler kornea lamellerine de nüfuz eder ve kollajen lifler ile proteoglikanlarda dejenerasyona neden olur.
TACSTD2 geninde 20’den fazla mutasyon bildirilmiştir12). Japonya’da Q118X mutasyonu (anlamsız mutasyon, fonksiyonel null) kurucu mutasyon olarak hastalık kromozomlarının %80’inden fazlasını oluşturur2). Hastalık tipik olarak homozigot bireylerde ortaya çıkar, ancak farklı aileler arasındaki evliliklerden kaynaklanan bileşik heterozigotlarda da görülür. İlginç bir şekilde, aynı Q118X homozigot mutasyonuna sahip bireylerde bile dört klinik tip (dut benzeri, bant benzeri, kamkat benzeri ve parankimal opasite) karışık olarak gözlenir3).
Kornea amiloidozu, primer veya sekonder, sistemik veya lokal olmasına göre sınıflandırılır. GDLD, latis kornea distrofisi ile birlikte primer lokal amiloidoz grubunda yer alır. Sekonder lokal amiloid dejenerasyon, trikiyazis, keratokonus, travma, uzun süreli kontakt lens kullanımı gibi durumlara bağlı olarak gelişir ve ayırıcı tanıda düşünülmelidir.
GDLD nadir bir hastalıktır ve bireysel merkezlerde klinik deneyime sahip hekim sayısı azdır; standart tanı ve tedavi yöntemleri bulunmamaktaydı. Sağlık, Çalışma ve Refah Bakanlığı’nın İnatçı Hastalıklar Politika Araştırma Projesi kapsamındaki “Nadir İnatçı Kornea Hastalıkları Epidemiyolojik Araştırma Grubu” ve “Ön Segment Nadir Hastalıkları Klinik Kılavuzu Oluşturma ve Yaygınlaştırma Araştırma Grubu” tarafından tanı kriterleri ve şiddet sınıflaması oluşturulmuştur2). 2019 yılında “Jelatinöz Damla Benzeri Kornea Distrofisi” olarak nadir hastalık statüsü almış ve halen Minds (Medical Information Network Distribution Service) uyumlu klinik kılavuz hazırlanmaktadır2).
Uzun vadede nüks ve tekrarlayan tedaviler sorun oluşturur. Terapötik kontakt lens ve limbal transplantasyonun birlikte kullanımı nüksü geciktirebilir ve cerrahi aralıkları uzatarak uzun dönem prognozu iyileştirebilir6, 7, 10).
Maeno ve ark., sadece bir gözde tekrarlayan amiloid birikimi gösteren klinik olarak atipik bir olgu bildirerek GDLD’nin fenotipik çeşitliliğini ortaya koymuştur13). Bu tür atipik olguların tanısında TACSTD2 gen testi belirleyici rol oynar2).
Temel araştırmalarda, TACSTD2’nin aşağı akışındaki sıkı bağlantı molekül dinamiklerinin ayrıntılı olarak aydınlatılması ve Claudin stabilizasyonunu hedefleyen tedavilerin geliştirilmesi beklenmektedir. Klinik açıdan, yapay kornea, kornea epitel tabakası nakli ve iPS hücre kaynaklı kornea epiteli gibi rejeneratif tıp yaklaşımlarının endikasyonlarının genişletilmesi değerlendirilmektedir.