Nedeni, TGFBI genindeki (kromozom 5q31) nokta mutasyonudur ve otozomal dominant kalıtım gösterir. Mutasyon farklılığına göre aşağıdaki iki tipe ayrılır.
Sınıflandırma
Ana mutasyon
Diğer adı/eski adı
Ana birikintiler
GCD1
Arg555Trp (R555W)
Klasik granüler, Groenouw tip 1
Sadece hiyalin
GCD2
Arg124His (R124H)
Avellino kornea distrofisi
Hyalin + amiloid
GCD2, 1988 yılında GCD1’den bağımsız bir alt tip olarak rapor edilmiş ve ilk ailenin İtalya’nın Avellino bölgesinden gelmesi nedeniyle Avellino kornea distrofisi olarak adlandırılmıştır 1). Daha sonra 1997 yılında sorumlu gen TGFBI tanımlanmış ve R124H mutasyonu belirlenmiştir 3). IC3D sınıflandırmasının 2. baskısından (2015) itibaren resmi olarak granüler kornea distrofisi tip 2 (GCD2) olarak adlandırılmış ve ‘Avellino’ tarihsel isim olarak parantez içinde belirtilmektedir 2).
Granüler kornea distrofisi, uluslararası olarak epitelyal-stromal distrofiler grubunda sınıflandırılır. Bu grup, TGFBI ile ilişkili granüler (GCD1 ve GCD2), latis (LCD1 ve LCD3A), Reis-Bücklers ve Thiel-Behnke olmak üzere altı hastalığı içerir ve hepsi aynı 5q31 kromozomundaki TGFBI genindeki farklı nokta mutasyonlarından kaynaklanır 2,4). Japonya’daki oftalmoloji pratiğinde, bu altı hastalık topluca ‘TGFBI ilişkili kornea distrofileri’ olarak adlandırılır.
Granüler kornea distrofisi ilk kez 1890 yılında Groenouw tarafından rapor edilmiş ve o zamanlar sadece ‘Groenouw tip 1’ olarak adlandırılmıştır. 1938 yılında latis distrofiden ayrımı netleşmiş ve uzun süre ‘granüler kornea distrofisi’ olarak tek bir hastalık olarak ele alınmıştır. 1988 yılında İtalya’nın Avellino bölgesinden bir ailede hem granüler hem de latis özellikler gösteren bir form rapor edilmiş ve bu daha sonra GCD2 (Avellino tipi) olarak ayrılmıştır 1,2). 2015 yılındaki IC3D sınıflandırma revizyonu ile mevcut sınıflandırma sistemi oluşturulmuş ve genotip temelli adlandırma uluslararası standart haline gelmiştir 2).
Kalıtım şekli: Otozomal dominant, yüksek penetrans
Tip 1: Avrupa ve Amerika’da sık, Japonya’da nadir
Tip 2: Japonya, Kore gibi Doğu Asya’da çok sık. Kore’de prevalans yaklaşık 10.000’de 11.5 kişi 1)
TGFBI ilişkili kornea distrofileri içindeki oranı: GCD2, Kore ve Japonya’da %72-91, ABD’de %36, Polonya’da %3 1)
Japonya’da genetik tanı verileri: Yamaguchi Üniversitesi’nde 2000-2021 yılları arasında 21 yılda 234 kornea distrofisi hastasına genetik tanı konulmuş ve dört ana kornea distrofisi (granüler tip I ve II, latis tip I ve IIIA, kolloid damla, maküler) tüm vakaların yaklaşık %96’sını oluşturmuştur. - Doğu Asya’da özellik: Granüler kornea distrofisi Doğu Asya’da ağırlıklı olarak tip 2 (R124H) görülür. - Başlangıç yaşı: Heterozigot GCD2’de okul çağından itibaren sadece yarık lambada görülebilen mikroskobik bulanıklıklar vardır ancak subjektif semptom yoktur. Subjektif görme azalması tipik olarak 40-50’li yaşlarda ortaya çıkar.
Cinsiyet farkı: Otozomal dominant kalıtım olduğu için cinsiyet farkı yoktur.
Q«Granüler» hangi durumu ifade eder?
A
Kornea merkezinin yüzeyel stromasında, sınırları belirgin beyaz ila grimsi beyaz küçük kümeler (granüler birikintiler) oluşması durumunu ifade eder. Yarık lamba mikroskobu ile doğrudan gözlemlendiğinde ekmek kırıntısı, kar tanesi veya akide şekeri olarak tanımlanır. Birikintiler mutant TGFBI proteininden kaynaklanır.
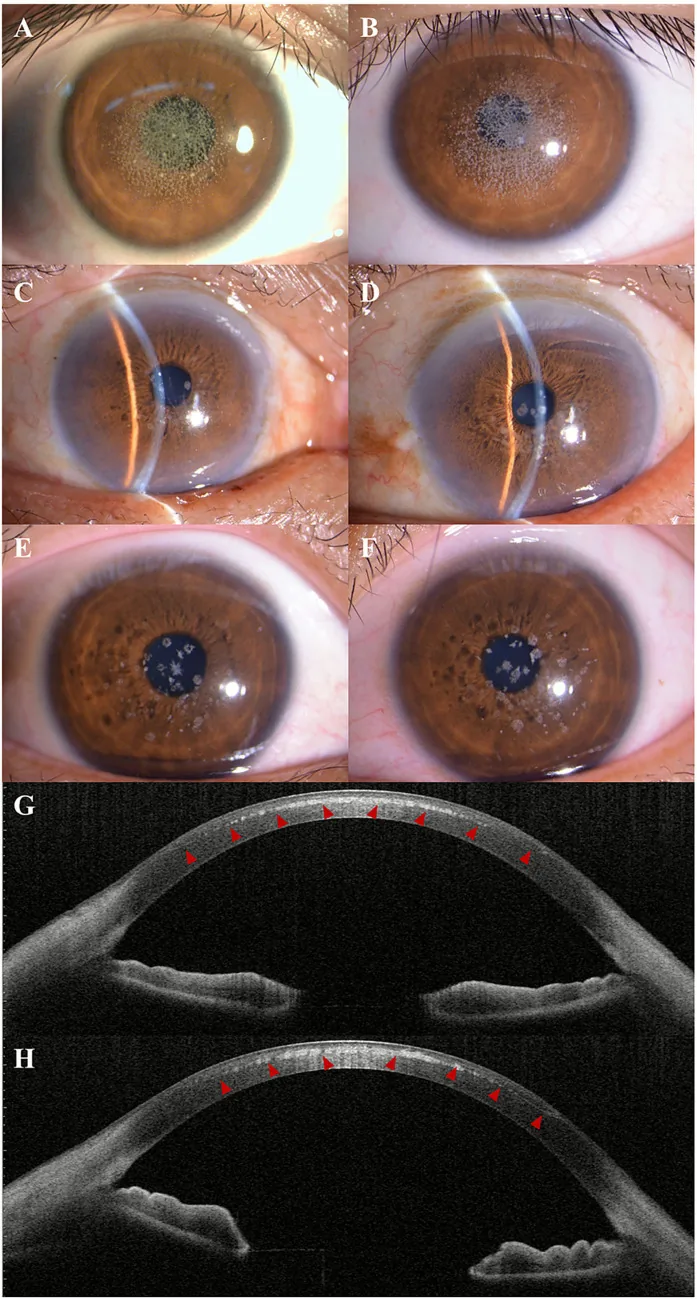
Kuang L, et al. Case Report: Post-LASIK exacerbation of granular corneal dystrophy type 2: a familial case with TGFBI mutation. Front Med (Lausanne). 2025. Figure 1. PMCID: PMC12722859. License: CC BY.
Yarık lamba fotoğrafında, kornea merkezinden parasantrale grimsi beyaz granüler bulanıklıklar dağınık ve kümelenmiş halde görülmektedir. AS-OCT’de ön stromada yüksek yansıtıcı birikintiler izlenmekte olup granüler kornea distrofisinin klinik bulgusunu göstermektedir.
Asemptomatik ila hafif: Heterozigotlarda erken-orta evrelerde görme azalması hissedilmez ve sıklıkla taramalarda tesadüfen keşfedilir. Çoğu hasta 40-50’li yaşlarda görme azalmasından şikayet eder.
Kamaşma ve fotofobi: Bulanıklıklar pupil alanına uzandığında gündüz kamaşması ve kontrast duyarlılığında azalma şikayeti olur.
Tekrarlayan kornea epitel erozyonu: Birikintiler Bowman tabakası ve epitel bazal membranına zarar vererek uyku sırasında veya uyanırken keskin göz ağrısı, sulanma ve kızarıklığa neden olur.
Görme azalması: Birikintiler arasındaki şeffaf alanlar opaklaştığında görme ilerleyici şekilde azalır3).
Kontrast duyarlılığında azalma: Görme keskinliğinden (Landolt halkası testi) önce kontrast duyarlılığı sıklıkla azalır.
Gündüz körlüğü eğilimi: Aydınlık ortamda saçılan ışıktan güçlü etkilenme nedeniyle dışarıda veya araba kullanırken kamaşma şikayeti olur.
Gözlük veya kontakt lensle düzeltilememe: Birikintilerin saçılması refraktif düzeltme ile iyileşmez.
Homozigot bireylerde çocukluk döneminde (4-7 yaş) belirgin görme azalması başlar ve yaklaşık 10 yaşında tedavi gerektirir.
Granüler opasiteler: Kornea merkezinde sınırları belirgin, nispeten küçük, beyaz ila grimsi beyaz granüler opasiteler dağınık halde bulunur. Ekmek kırıntısı veya kar tanesi şeklinde tanımlanır.
Derinlik: Kornea epiteli altı ve stromanın yüzeyel tabakaları. Limbusa ulaşmaz.
Biriken madde: Yalnızca hiyalin. Masson trikrom boyası ile kırmızı boyanır. Amiloid içermez.
İlerleme: Yaşla birlikte granül sayısı artar ve sınırlar belirsizleşir.
GCD2 (R124H)
Granüler opasiteler: GCD1’den daha büyük, beyaz ila grimsi beyaz, sınırları belirgin opasitelerle başlar. Fenotipler çeşitlidir: akide şekeri, çizgisel, yıldız, sopa şeklinde vb.
Karma tip: Bazen latis distrofisinde görülen ağsı ince çizgisel opasiteler de görülebilir.
Biriken madde: Hem hiyalin hem amiloid. Masson trikrom pozitif ve Kongo kırmızısı pozitif, polarizasyon mikroskobunda sarı-yeşil renk alır.
İlerleme: 25-30 yaş üzerinde stroma orta tabakasında sopa ve yıldız şeklinde yoğun beyaz opasiteler eklenir. Yüzeyel tabakada yaygın plak benzeri birikim artar ve PTK iyi bir endikasyondur3).
Her iki tipte de opasiteler kornea merkezinde yer alır ve limbus çevresine uzanmaz. Genellikle iki taraflıdır ve iki göz arasında fark azdır.
Homozigot mutasyonlarda fenotip belirgin şekilde farklıdır.
GCD1 homozigot: Kornea epiteli altından stroma yüzeyel tabakalarına kadar aynı derinlikte, neredeyse boşluksuz beyaz retiküler opasiteler bulunur. İlerlediğinde iris ve ön kamara gözlenemez hale gelir.
GCD2 homozigot: En periferik kısım hariç tüm kornea yüzeyinde boşluksuz, yoğun yuvarlak beyaz opasiteler oluşur. Çıplak gözle bile beyazlık fark edilecek kadar şiddetlidir ve sadece limbal şeffaflık korunur3). Homozigot vakalar, PTK veya kornea naklinden sonra bile 1-2 yıl gibi kısa sürede nükseden, tedaviye dirençli kornea distrofileridir.
QHomozigot ve heterozigotlarda seyir nasıl farklılık gösterir?
A
Homozigotlar çocukluk döneminde (4-7 yaş) başlar ve hızlı ilerler. Tüm kornea yüzeyinde boşluksuz beyaz opasiteler oluşur ve 10 yaş civarında PTK veya kornea nakli gerekir. Ameliyat sonrası 1-2 yıl içinde nükseder ve tedaviye dirençli bir seyir izler. Heterozigotlar yavaş ilerler ve genellikle 40-50’li yaşlara kadar iyi görme korunur.
GCD, TGFBI genindeki (kromozom 5q31) nokta mutasyonları sonucu oluşur. TGFBI geni, hücre dışı matriks proteini TGFBIp (keratoepitelin) kodlar. Mutant TGFBIp, protein yıkımına karşı duyarlılığı azalmıştır ve kornea stromasında anormal çözünmeyen birikintiler olarak birikir1,5,7).
TGFBI ile ilişkili kornea distrofileri şunları içerir2,4):
Granüler kornea distrofisi tip 1 (R555W)
Granüler kornea distrofisi tip 2 (R124H, eski adı Avellino)
Homozigot: Aynı mutasyonun homozigot olduğu durumlarda daha şiddetli fenotip görülür
Kornea cerrahisi: GCD2, kornea travması sonrası hızla ilerleyebilir. Özellikle lazer refraktif cerrahi sonrası bulanıklık belirgin şekilde kötüleşir1,8,9)
Irk: GCD2, Doğu Asya’da (Kore, Japonya) daha sıktır. Genetik kurucu etkisinin (founder effect) rol oynadığı belirtilmektedir3)
Çevresel faktörler bilinmiyor: UV maruziyeti veya diyabetin doğrudan etkisi şu anda kanıtlanmamıştır
GCD, yüksek penetranslı otozomal dominant bir hastalıktır. Probanda kesin tanı konulduğunda, birinci derece akrabaların (ebeveyn, kardeş, çocuk) %50’si aynı mutasyonu taşıyabilir. Ailedeki semptomsuz taşıyıcıların erken tespiti, gelecekte refraktif cerrahiden kaçınmayı ve ilerlemeyi izlemek için düzenli muayene planlamasını sağlayabilir1,5). Özellikle LASIK isteyen gençlerde, ayrıntılı aile öyküsü alınması ve gerektiğinde genetik test yapılması şiddetle önerilir.
Klinik tanı, yarık lamba ile ön stromada sınırları belirgin granüler bulanıklıkların gözlenmesi ve pozitif aile öyküsüne dayanır. Hiperemi ve kornea ödemi olmaksızın iki taraflı kornea bulanıklığı (birikinti) varsa kornea distrofisinden şüphelenilir3).
Kornea distrofilerinin ayırıcı tanısında, önce birikintilerin “sınırları belirgin” mi yoksa “yaygın” mı olduğu belirlenir3). Sınırları belirgin granüler birikintiler varsa, birikinti boyutuna göre GCD1 (küçük) ve GCD2 (büyük) ayırt edilir. GCD2’de skleral saçılma yöntemiyle granüler birikintiler arasında yaygın yüzeysel bulanıklık görülebilir ve bu yüzeysel bulanıklık PTK için iyi bir endikasyondur3).
Yarık lamba: Sınırları belirgin beyaz granüler bulanıklıkların doğrudan gözlenmesi. Skleral saçılma, retroillüminasyon ve transillüminasyon yöntemleri de kullanılır
Ön Segment Optik Koherens Tomografi (AS-OCT): Ön stromada yüksek yansıtıcılı opasiteleri gösterir. PTK için eksizyon derinliği planlamasında faydalıdır.
Konfokal Mikroskopi: Epitel-Bowman tabakası arasında düzensiz, yüksek yansıtıcılı, ekmek kırıntısı benzeri opasiteler görülür.
Ultrasonik Biyomikroskopi (UBM): Yüzeyel stromada yüksek yansıtıcılı granülleri tespit eder.
Kornea Topografisi Analizi: Opasitenin yoğunluğu hakkında ek bilgi sağlar.
Genetik Test: TGFBI gen analizi kesin tanı için faydalıdır. Japonya’da Nisan 2020’den itibaren kornea distrofisi genetik testi olarak sigorta kapsamına alınmıştır 3).
Ön Segment Fotoğraflama: Uzun süreli takip için ilk muayenede ve her takipte yüksek kaliteli ön segment fotoğrafları çekilip tıbbi kayıtlara eklenmesi önemlidir.
GCD1: Masson trikrom boyası ile kırmızı boyanan hyalin birikintileri. Amiloid içermez. Elektron mikroskopisinde çubuk veya yamuk şeklinde birikintiler.
GCD2: Hem hyalin (Masson trikrom pozitif) hem de amiloid (Kongo kırmızısı pozitif, polarize mikroskopta sarı-yeşil) birikir. Elektron mikroskopisinde çubuk şeklinde elektron yoğun birikintiler ve amiloid fibrilleri görülür 1,7).
Kafes benzeri kornea distrofisi tip 1 (LCD1): TGFBI R124C mutasyonu. Stromada amiloid birikimine bağlı lineer ve kafes benzeri opasiteler. Tekrarlayan kornea epitel erozyonlarına yatkınlık 3)
Benekli kornea distrofisi (FCD): PIP5K3 mutasyonu. Stromada küçük beyaz noktalar, genellikle asemptomatik
QGenetik test sigorta kapsamında mı?
A
Nisan 2020’den itibaren kornea distrofisi genetik testi sigorta kapsamına alınmıştır. Ancak tesis onayı gerektiğinden testi yapabilen merkezler sınırlıdır. Klinik bulgular şüpheliyse veya LASIK gibi refraktif cerrahi düşünülüyorsa genetik test ile kesin tanı önerilir.
Görme azalması veya tekrarlayan epitel erozyonu olmayan erken evrelerde tedavi gerekmez. Görme azalması pupil alanına ulaştığında cerrahi müdahale düşünülür.
Yapay gözyaşı: Sodyum hyaluronat %0.1 veya %0.3 damla, günde 4-6 kez, kuruluk ve tahrişi azaltmak için
Terapötik yumuşak kontakt lens: Tekrarlayan epitel erozyonlarında göz yüzeyini korur ve iyileşmeyi hızlandırır. Tam gün kullanım ve düzenli değişim gereklidir
Antibiyotik damla ve merhem: Epitel erozyonu sırasında sekonder enfeksiyonu önlemek için, levofloksasin %0.5 damla günde 3-4 kez, yatmadan önce ofloksasin merhem
Hipertonik tuzlu su (%5 sodyum klorür damla/göz merhemi): Epitel ödemini azaltmak için yardımcı tedavi olarak kullanılabilir.
GCD2 homozigot: İlk PTK’dan yaklaşık 18 ay sonra nüks görülür; ikinci ve üçüncü PTK’dan sonra ise yaklaşık 3 ay içinde nüks eder1)
GCD2 heterozigot: PTK sonrası nüks ortalama 38,4 ay ile nispeten yavaştır1)
Mitomisin C (MMC) ile birlikte kullanım: PTK sırasında MMC kullanımı önerilmez. Çünkü MMC, kornea stromasındaki keratositlerin apoptozuna neden olur ve TGFBIp’nin geri emilim ve parçalanmasından sorumlu hücreleri azaltarak nüksü hızlandırabilir1)
LASIK sonrası kötüleşen vakalar: PTK uygulanabilir, ancak LASIK flebi kaldırıldıktan sonra etkinlik daha yüksektir1,8)
Ameliyat sonrası bakım: PTK sonrası epitel iyileşene kadar antibiyotikli göz damlası (levofloksasin %0,5) ve kortikosteroid göz damlası (fluorometolon %0,1) günde 4 kez, ardından azaltılarak kullanılır. Epitel iyileşmesi genellikle 3-5 gün sürer
DALK, endoteli koruyan bir yöntemdir. Büyük kabarcık tekniği (big bubble technique) kullanılarak stroma Descemet membranının hemen üzerine kadar ayrılır ve çıkarılır, ardından donör stroması dikilir. Endotel reddi riski olmadığından uzun dönem prognoz PK’den daha iyidir3). Kitazawa ve arkadaşlarının çalışmasında, TGFBI ile ilişkili kornea distrofilerinde (granüler ve latis tip dahil) DALK sonrası 5 yıllık görme genellikle iyiydi ve greft sağkalım oranı yüksekti10). Japonya’da bu yöntem sağlık sigortası kapsamında uygulanabilir.
GCD için cerrahi tedavi seçimi aşağıdaki sırayla değerlendirilir1,6):
Birikim ön stroma ile sınırlı (kornea epitel altı ~150 μm) → PTK
Birden fazla PTK sonrası nüks veya derin tabakalarda (150-400 μm) bulanıklık → DALK
Tüm katmanları tutan bulanıklık, endotel hasarı eşlik ediyorsa veya DALK uygulanamıyorsa → PK
Herhangi bir cerrahi yöntem sonrası tekrarlayan nüksler (özellikle GCD2 homozigot) → gen tedavisi gibi araştırma amaçlı yaklaşımların değerlendirilmesi
GCD için LASIK, LASEK, PRK ve SMILE’in tümü kontrendikedir. Ameliyat sonrası korneal opasite hızla kötüleşir ve ciddi görme azalmasına yol açar 1,8,9). LASIK sonrası flep ile stroma yatağı arasında çok sayıda küçük granüler birikim oluşur. LASIK, PRK’ya kıyasla daha şiddetli kötüleşmeye ve daha kötü nihai görme keskinliğine neden olur 1,8). Kore ve Japonya’dan gelen vaka raporları, ameliyat öncesi asemptomatik olan hastaların LASIK’ten aylar veya yıllar sonra belirgin korneal opasite geliştirdiğini ve PTK veya kornea nakli gerektirdiğini bildirmiştir 8,9).
QLASIK yaptırdıktan sonra GCD teşhisi konulursa ne olur?
A
LASIK sonrası GCD gelişen vakalarda, flep ile stroma yatağı arasında hızla granüler birikim oluşur. Tedavi seçenekleri arasında LASIK flebinin çıkarılmasını takiben PTK, DALK ve PK yer alır ve bu sırayla değerlendirilir. Erken dönemde bir göz uzmanına başvurmak önemlidir.
TGFBI geni, hücre dışı matriks proteini olan TGFBIp’yi (keratoepitelin, 68 kDa) kodlar. TGFBIp, hücre adezyonu, göçü ve çoğalmasında rol oynar ve normal kornea stromasında da eksprese edilir 1,5,7). TGFBI geninde mutasyon meydana geldiğinde, mutant TGFBIp proteolitik bozulmaya karşı duyarlılığı azalır ve kornea stromasında çözünmeyen birikintiler olarak birikir 5,7).
Mitokondriyal işlev bozukluğu: Mutant TGFBIp’nin kendisinin kornea fibroblastlarını etkileyerek mitokondriyal işlev bozukluğuna neden olabileceği öne sürülmüştür 1)
Korneal neovaskülarizasyonun etkisi: Korneal neovaskülarizasyonun eşlik ettiği bölgelerde birikintiler azalma ve yeniden emilme eğilimindedir. Bu bulgu, damar desteği olmayan kornea merkezinde birikintilerin yoğunlaşma mekanizmasını desteklemektedir1)
LASIK sonrası flep ve stroma yatağı arasında TGFBIp hızla birikir. Bunun, kornea merkezindeki cerrahi manipülasyonun mutant TGFBIp birikimini hızlandırmasından kaynaklandığı düşünülmektedir1,8). Katarakt cerrahisi sırasında yapılan kornea kesisi (limbus yakınında) kötüleşmeye yol açmadığından, vaskülarize limbus ile mesafenin ilişkili olduğu tahmin edilmektedir1). Awwad ve ark.‘nın patolojik gözlemleri, LASIK sonrası oluşan birikintilerin, flep ve stroma yatağı ara yüzündeki yara iyileşme reaksiyonuna eşlik eden keratosit aktivasyonu ile birlikte TGFBIp birikimini gösterdiğini düşündürmektedir8).
GCD1 birikintileri ışık mikroskobunda homojen eozinofilik maddeler olarak gözlenir ve Masson trikrom boyası ile kırmızı renkte boyanır. Elektron mikroskobu düzeyinde, 100-500 nm çapında çubuk veya yamuk şeklinde yüksek elektron yoğunluklu yapılar olarak tanınır6).
GCD2’de hyalin birikintilerine ek olarak amiloid fibrilleri (8-10 nm çapında) bulunur. Amiloid fibrilleri Kongo kırmızısı boyamasıyla turuncu-kırmızı renk alır ve polarizasyon mikroskobunda elma yeşili çift kırılım gösterir6). Çift boyama özelliği, GCD2’nin patolojik kesin tanısında faydalıdır.
Poulsen ve ark.‘nın proteomik analizine göre, GCD2 hastalarının korneasında mutant R124H-TGFBIp, normal TGFBIp’e kıyasla proteolitik enzimler tarafından kesilmeye daha dirençlidir ve belirli C-terminal fragmanları seçici olarak birikir6). Bu kesilme direncinin hyalin ve amiloid fibril oluşumunun temelini oluşturabileceği düşünülmektedir.
Lityum klorürün TGFBI protein üretimini azalttığı bildirilmiştir. Melatonin ve rapamisin kombinasyon tedavisinin TGFBI protein ekspresyonunu inhibe ederken aynı zamanda otofajiyi aktive ederek mutant TGFBIp’nin parçalanmasını hızlandırabileceği gösterilmiştir1,5).
Küçük enterferans yapan RNA (siRNA) veya kısa saç tokası siRNA (shRNA) kullanılarak mutant TGFBI ekspresyonunun susturulması preklinik olarak araştırılmaktadır. CRISPR/Cas9 ile genom düzenleme teknolojisi de bir adaydır, ancak normal alel veya diğer genler üzerinde istenmeyen hedef dışı etkiler bir zorluktur1,5).
Korneal elektroliz: Kornea nakli sonrası nüks vakalarında deneysel kullanım rapor edilmiştir. Uzun dönem sonuçları bilinmemektedir.
Makine öğrenimi ile tanı desteği: Ön segment fotoğraflarından GCD’yi otomatik olarak tanımlayan bir yapay zeka modelinin geliştirildiği bildirilmiştir.
Şaperon tedavisi: Mutant TGFBIp’nin doğru katlanmasına yardımcı olan kimyasal şaperonlar (4-fenilbütirik asit gibi) üzerine temel araştırmalar devam etmektedir3).
iPS hücre kaynaklı korneal epitel tabakası: Hastadan alınan iPS hücrelerinden üretilen korneal epitel tabakasının transplantasyon tedavisi, gelecekteki bir seçenek olarak araştırılmaktadır.
Ülke içinde, Japon Oftalmoloji Derneği ve Japon Kornea Derneği öncülüğünde TGFBI ile ilişkili korneal distrofiler için ulusal bir kayıt oluşturulmasına yönelik tartışmalar devam etmektedir. 2020 yılında genetik testin sigorta kapsamına alınmasıyla genetik tanı vakaları artmış ve Japonlara özgü mutasyon paternleri ve fenotiplerine ilişkin uzun dönem takip verileri birikmektedir3,11). Gelecekte, taşıyıcı taramasından refraktif cerrahi uygunluğuna kadar kanıta dayalı bir klinik sistemin oluşturulması beklenmektedir.
GCD hastalarında aşağıdaki yaşam tarzı önerileri önemlidir:
UV koruması: Dışarı çıkarken UV korumalı güneş gözlüğü veya gözlük kullanarak korneanın UV’ye maruziyetini azaltın.
Göz travmasından kaçınma: Darbe veya yabancı cisim kaynaklı kornea hasarı, tekrarlayan epitel erozyonuna yol açabilir. Spor veya çalışma sırasında koruyucu gözlük kullanın.
Düzenli kontroller: Heterozigot bireylerde bile yılda bir kez yarık lamba muayenesi ve görme testi yaptırın.
Aile bireylerinin taranması: Birinci derece yakınlara aile içi tarama önerin.
Kontakt lensler: Yumuşak lensler genellikle mümkündür, ancak kullanım süresini kısa tutun ve düzenli değişim yapın.
Refraktif cerrahiden kaçınma: LASIK tipi cerrahiler kesinlikle kontrendikedir. Gözlük veya kontakt lens ile düzeltmeyi tercih edin.
Chang MS, Jun I, Kim EK. Mini-Review: Clinical Features and Management of Granular Corneal Dystrophy Type 2. Korean journal of ophthalmology : KJO. 2023;37(4):340-347. doi:10.3341/kjo.2023.0032. PMID:37336511; PMCID:PMC10427907.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Lisch W, Weiss JS. Clinical and genetic update of corneal dystrophies. Experimental eye research. 2019;186:107715. doi:10.1016/j.exer.2019.107715. PMID:31301286.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern®. Ophthalmology. 2024;131(4):P1-P79.
Poulsen ET, Dyrlund TF, Runager K, et al. Proteomics of granular corneal dystrophy type 2 reveals altered TGFBIp proteolytic processing. J Proteome Res. 2014;13(10):4659-4667.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Awwad ST, Di Pascuale MA, Hogan RN, Forstot SL, McCulley JP, Cavanagh HD. Avellino corneal dystrophy worsening after laser in situ keratomileusis: further clinicopathologic observations and proposed pathogenesis. American journal of ophthalmology. 2008;145(4):656-61. doi:10.1016/j.ajo.2007.12.008. PMID:18243154; PMCID:PMC2661203.
Jun RM, Tchah H, Kim TI, et al. Avellino corneal dystrophy after LASIK. Ophthalmology. 2004;111(3):463-468.
Kitazawa K, Kawasaki S, Shinomiya K, et al. Safety of anterior lamellar keratoplasty for recurrent TGFBI corneal dystrophy. Br J Ophthalmol. 2016;100(4):448-454.