这是一种由TGFBI基因突变引起的常染色体显性遗传 性角膜营养不良 ,导致角膜基质 中透明质和淀粉样蛋白沉积。
分为1型(R555W突变,Groenouw 1型)和2型(R124H突变,旧称Avellino角膜营养不良 )。
在日本,2型占绝大多数。韩国的患病率约为每1万人中11.5人1) 。
根据山口大学的基因诊断研究,四大角膜营养不良 (颗粒状I/II型、格子状I/IIIA型、胶滴状、斑状)约占所有病例的96%。- 纯合子患者在儿童早期(4-7岁)发病且病情严重。杂合子患者进展缓慢。
PTK (治疗性角膜切除术 )是首选治疗。对于深层混浊,选择DALK 。LASIK 、PRK、LASEK、SMILE 等激光屈光 矫正手术为禁忌。 颗粒状角膜营养不良(GCD)是一种遗传性角膜 疾病,特征为角膜基质 中出现颗粒状沉积物。在国际角膜营养不良 分类第二版(IC3D 2015)中,它被归类为上皮-基质TGFBI相关性营养不良2) 。
该病由TGFBI基因(染色体5q31)的点突变引起,呈常染色体显性遗传 。根据突变不同,分为以下两型。
分类 主要突变 别名/旧称 主要沉积物 GCD1 Arg555Trp (R555W) 经典颗粒状 / Groenouw 1型 仅透明质 GCD2 Arg124His(R124H) 阿韦利诺角膜营养不良 透明蛋白+淀粉样蛋白
GCD2于1988年被报道为独立于GCD1的亚型,因首个家系源自意大利阿韦利诺地区,故称为阿韦利诺角膜营养不良 1) 。随后在1997年,致病基因TGFBI被确定,并鉴定出R124H突变3) 。自IC3D分类第二版(2015年)起,正式命名为颗粒状角膜营养不良2型(GCD2),而“阿韦利诺”作为历史名称并列标注2) 。
颗粒状角膜营养不良在国际上被归类于上皮-基质营养不良组。该组包括六种TGFBI相关疾病:颗粒状(GCD1、GCD2)、格子状(LCD1、LCD3A)、Reis-Bücklers和Thiel-Behnke,均由5q31上TGFBI基因的不同点突变引起2,4) 。在日本眼科临床中,这六种疾病统称为“TGFBI相关角膜营养不良 ”。
颗粒状角膜营养不良于1890年由Groenouw首次报道,当时仅称为“Groenouw 1型”。1938年与格子状营养不良明确区分,长期作为单一疾病“颗粒状角膜营养不良”处理。1988年,意大利阿韦利诺地区的一个家系报告了兼具颗粒状和格子状特征的病型,后分离为GCD2(阿韦利诺型)1,2) 。2015年IC3D分类修订确立了现行分类体系,基于基因型的命名成为国际标准2) 。
遗传方式 :常染色体显性遗传 ,外显率高1型 :欧美多见,日本少见2型 :日本、韩国等东亚地区占绝对多数。韩国患病率约为每万人11.5人1) 在TGFBI相关角膜营养不良 中的占比 :GCD2在韩国和日本占72-91%,美国占36%,波兰占3%1) 日本基因诊断数据 :山口大学在2000年至2021年的21年间对234例角膜营养不良 患者进行了基因诊断,四大角膜营养不良 (颗粒状I型、II型,格子状I型、IIIA型,胶滴状,斑状)约占全部病例的96%。- 东亚特征 :颗粒状角膜营养不良在东亚以2型(R124H)为主。- 发病年龄 :杂合子GCD2从学龄期起仅在裂隙灯 下可见微小混浊,但无自觉症状。自觉视力 下降通常出现在40~50岁。性别差异 :常染色体显性遗传 ,无性别差异。
Q
“颗粒状”指的是什么状态?
A
指角膜 中央部实质浅层出现许多边界清晰的白色至灰白色小块(颗粒状沉积物)。用裂隙灯 显微镜直接观察时,形容为面包屑状、雪花状或金平糖状。沉积物来源于突变的TGFBI蛋白。
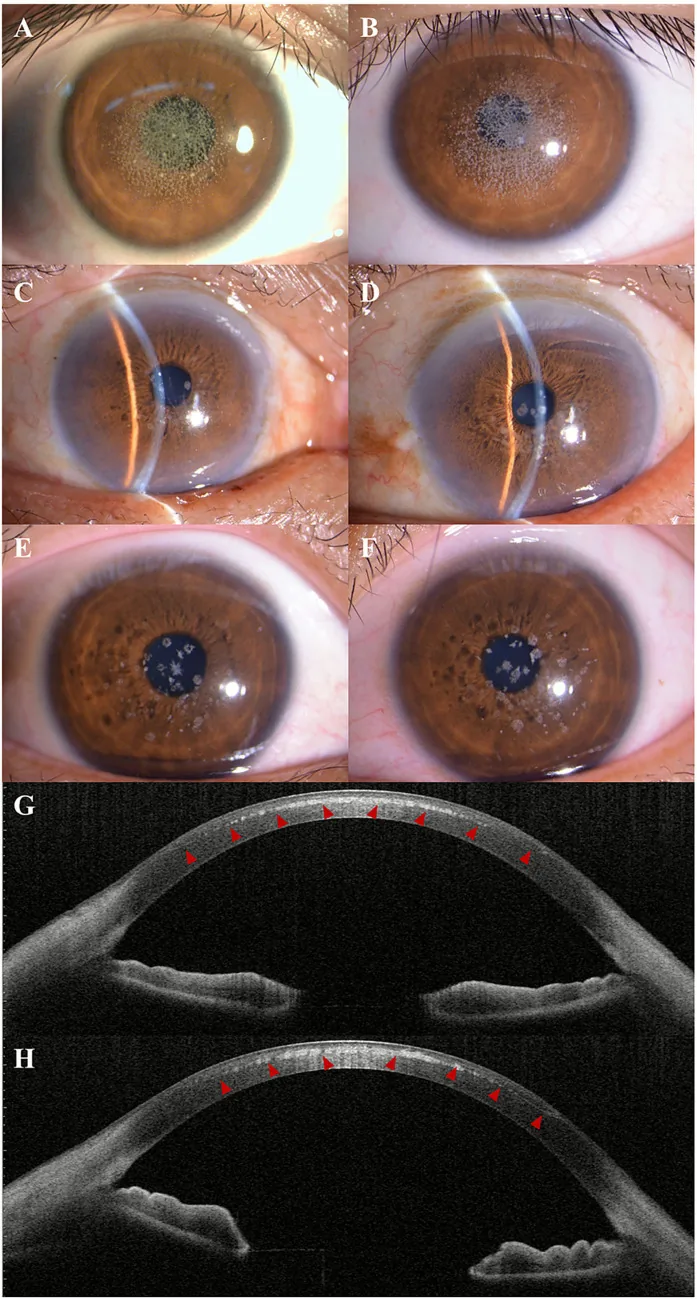
Kuang L, et al. Case Report: Post-LAS
IK exacerbation of granular corneal dystrophy type 2: a familial case with TGFBI mutation. Front Med (Lausanne). 2025. Figure 1. PM
CI D: PMC12722859. License: CC BY.
裂隙灯 照片显示
角膜 中央至旁中央有散在或聚集的灰白色颗粒状混浊。
AS-OCT 显示
角膜 前部实质有高反射沉积物,呈现颗粒状角膜营养不良的临床所见。
无症状至轻度 :杂合子早期至中期常无自觉视力 下降,不少病例在体检中偶然发现。多数在40~50岁后主诉视力 下降。眩光、畏光 :混浊波及瞳孔 区时,主诉白天眩光和对比敏感度 下降。复发性角膜上皮糜烂 :沉积物损伤鲍曼层和上皮基底膜,睡眠中或起床时出现剧烈眼痛 、流泪、充血 。视力 下降视力 进行性下降3) 。对比敏感度 下降对比敏感度 常先于视力 (兰多尔特环检查)下降。昼盲倾向 :明亮环境下散射光影响强烈,因此在户外或驾驶时主诉眩光。眼镜或隐形眼镜难以矫正 :沉积物的散射无法通过屈光 矫正改善。
纯合子患者从幼年(4-7岁)开始出现明显的视力 下降,大约在10岁前后需要治疗。
GCD1(R555W)
颗粒状混浊 :角膜 中央部散在边界清晰、相对较小的白色至灰白色颗粒状混浊。描述为面包屑状或雪片状。
深度 :角膜上皮 下及角膜基质 浅层。不累及角膜缘 。
沉积物 :仅透明蛋白。Masson三色染色呈红色。不含淀粉样蛋白。
进展 :随着年龄增长,颗粒数量增加,边界变得不清晰。
GCD2(R124H)
颗粒状混浊 :发病时混浊比GCD1更大,呈白色至灰白色,边界清晰。表型多样,如金平糖状、线状、星状、棒状等。
混合型 :有时可见格子状角膜营养不良 中出现的网状细线状混浊。
沉积物 :同时含有透明蛋白和淀粉样蛋白。Masson三色染色阳性且刚果红染色阳性,偏振光显微镜下呈黄绿色。
进展 :25-30岁以后,基质中层出现棒状、星状的浓密白色混浊。浅层弥漫性片状沉积增强,是PTK 的良好适应症3) 。
两种类型的混浊均位于角膜 中央部,不累及角膜缘 周边部。通常为双眼性,左右差异较小。
纯合子突变的表现型显著不同。
GCD1纯合子 :角膜上皮 下至浅层基质同一深度,存在几乎无间隙的白色网状混浊。进展后虹膜 和前房 无法观察。GCD2纯合子 :除最周边部外,角膜 全面出现无间隙的实性圆形白色混浊。严重到肉眼可识别白色,仅角膜缘 透明性得以保留3) 。纯合子病例即使进行PTK 或角膜移植 后,也会在1-2年的短期内复发,属于难治性角膜营养不良 。
Q
纯合子和杂合子的病程有何不同?
A
纯合子在幼儿期(4-7岁)发病,进展迅速。角膜 全面出现无间隙的白色混浊,10岁左右需要PTK 或角膜移植 。术后1-2年复发,呈难治性病程。杂合子缓慢进展,通常到40-50岁仍能维持良好视力 。
GCD由TGFBI基因(染色体5q31)的点突变引起。TGFBI基因编码细胞外基质蛋白TGFBIp(角膜上皮 素)。突变TGFBIp对蛋白水解的敏感性降低,在角膜基质 内作为异常不溶性沉积物积累1,5,7) 。
TGFBI相关角膜营养不良 包括以下类型2,4) :
家族史 :常染色体显性遗传 ,患者子女的50%有发病风险。纯合子 :同一突变的纯合子表现更严重的表型。角膜 手术角膜 外伤后可快速进展。尤其在激光屈光 矫正手术后混浊明显加重1,8,9) 。种族 :GCD2在东亚(韩国、日本)多见。有研究指出存在遗传奠基者效应3) 。环境因素未明 :紫外线暴露或糖尿病的直接影响目前尚未确定。
GCD是一种高外显率的常染色体显性遗传 病。若先证者确诊,其一级亲属(父母、兄弟姐妹、子女)有50%可能携带相同突变。早期识别家族中无症状携带者,有助于避免未来进行屈光 矫正手术,并制定定期随访计划以监测进展1,5) 。尤其对于考虑LASIK 的年轻人,强烈建议详细询问家族史并在必要时进行基因检测。
临床诊断基于裂隙灯 显微镜下观察到前基质层边界清晰的颗粒状混浊,以及阳性家族史。若出现双侧角膜 混浊(沉积物)而无充血 和角膜水肿 ,应怀疑角膜营养不良 3) 。
在角膜营养不良 的鉴别诊断中,首先判断沉积物是“边界清晰”还是“弥漫性”3) 。对于边界清晰的颗粒状沉积物,根据沉积物大小区分GCD1(小)和GCD2(大)。在GCD2中,巩膜 散射法可在颗粒状沉积物之间观察到弥漫性片状混浊,这种片状混浊是PTK 的良好适应证3) 。
裂隙灯 显微镜巩膜 散射法、后照法和透照法。眼前节光学相干断层扫描 (AS-OCT ) :显示前基质层的高反射混浊。有助于规划PTK 切除深度。共聚焦显微镜 Bowman层 之间可见不规则、高反射的面包屑样混浊。超声生物显微镜 (UBM )角膜地形图 基因检测 :TGFBI基因分析有助于确诊。在日本,自2020年4月起,作为角膜营养不良 基因检测纳入医保 3) 。眼前节照相 :为长期随访,初诊及每次随访时拍摄高质量眼前节照片并留存于病历中非常重要。
熟练的裂隙灯 检查中,结合以下观察方法可提高敏感性 3) 。
直接照明 :评估边界清晰的颗粒状沉积物的大小和密度。巩膜 散射法后部反光法 :利用通过瞳孔 区的光线检测微小沉积物。透照法 :早期发现上皮下轻微的颗粒状混浊。
GCD1 :Masson三色染色呈红色的透明样沉积物。不含淀粉样蛋白。电镜下呈杆状或梯形沉积物。GCD2 :同时沉积透明样物质(Masson三色染色阳性)和淀粉样蛋白(刚果红染色阳性,偏振光下呈黄绿色双折射)。电镜下可见杆状电子致密沉积物和淀粉样纤维 1,7) 。
Q
基因检测可以医保报销吗?
A
自2020年4月起,角膜营养不良 基因检测已纳入医保。但需要机构认证,因此可进行检测的机构有限。当临床发现可疑或考虑进行LASIK 等屈光 矫正手术时,建议通过基因检测确诊。
在视力 下降或复发性角膜上皮 脱落尚未出现的早期阶段,无需治疗。当视力 下降累及瞳孔 区时,考虑手术干预。
人工泪液 :使用0.1%或0.3%透明质酸钠滴眼液,每日4~6次,减轻干燥和刺激治疗性软性角膜 接触镜 :针对复发性上皮脱落,保护眼表并促进愈合。通常全天佩戴,需定期更换抗菌滴眼液/眼膏 :预防上皮脱落时的继发感染,使用0.5%左氧氟沙星滴眼液每日3~4次,睡前使用氧氟沙星眼膏高渗盐水(5%氯化钠滴眼液/眼膏) :可作为辅助治疗减轻上皮水肿。
根据沉积深度选择治疗方法。最终目标是尽可能推迟角膜移植 1,6) 。
PTK(治疗性角膜切除术)
适应症 :前基质混浊。推荐作为一线治疗1) 。
优点 :可重复进行。无移植排斥风险。上皮下弥漫性混浊是PTK 的良好适应症3) 。
操作 :准分子激光每次切除约50 μm基质。切除深度受角膜 厚度限制1,8) 。
局限性 :每次切除导致约1.5 D远视 。手术次数有限。
DALK/ALK(深板层角膜移植术)
适应症 :深基质混浊。当PTK 因角膜 厚度限制无法进行时。
优点 :由于角膜内皮 正常,DALK 更受青睐。无内皮型排斥反应风险3) 。
复发 :复发发生在宿主-移植物交界处或移植物浅层基质3) 。
PK(穿透性角膜移植术)
适应症 :反复复发导致深层基质沉积时的最后手段。
复发中位时间 :PK最长,为13.7年。PTK 、ALK和DALK 为2.7至3.7年1) 。
最终矫正视力 :所有术式效果相当(20/25至20/30)1) 。
GCD2纯合子 :首次PTK 后约18个月复发,第二次及以后约3个月复发1) 。GCD2杂合子 :PTK 后复发相对缓慢,平均38.4个月1) 。联合使用丝裂霉素C(MMC) :不推荐在PTK 时使用MMC。MMC诱导角膜基质 中角膜 细胞的凋亡,减少负责TGFBIp再吸收和降解的细胞,可能加速复发1) 。LASIK 后加重病例 :可行PTK ,但去除LASIK 瓣后效果更佳1,8) 。术后管理 :PTK 后使用抗菌滴眼液(左氧氟沙星0.5%)和皮质类固醇 滴眼液(氟米龙0.1%),每日四次直至上皮愈合,然后减量。上皮愈合通常需要3至5天。
DALK 是一种保留内皮的术式。采用大气泡技术,将基质剥离至紧贴后弹力层上方,缝合供体基质。由于没有内皮排斥反应的风险,长期预后优于PK3) 。Kitazawa等人的研究表明,TGFBI相关角膜营养不良 (包括颗粒状和格子状)患者DALK 术后5年视力 普遍良好,植片存活率高10) 。在日本,该手术可在医保下进行。
GCD的手术治疗选择按以下顺序考虑1,6) 。
沉积物局限于前部基质(上皮下至约150μm)→ PTK
多次PTK 后复发或深层混浊(150–400μm)→ DALK
全层混浊、合并内皮功能障碍或无法行DALK 的病例→ PK
任何术后反复复发的难治性病例(尤其是GCD2纯合子)→ 考虑基因治疗 等研究性方法
GCD是LASIK 、LASEK、PRK和SMILE 的禁忌症。术后角膜 混浊迅速加重,导致严重视力 下降1,8,9) 。LASIK 术后,角膜 瓣与基质床之间会形成大量小颗粒状沉积物。与PRK相比,LASIK 的病情加重更严重,最终视力 也更差1,8) 。来自韩国和日本的病例报告描述了许多术前无症状的患者,在LASIK 术后数月至数年内出现明显的角膜 混浊,需要PTK 或角膜移植 8,9) 。
Q
如果在接受LASIK后发现GCD,会怎样?
A
在LASIK 术后发生GCD的病例中,角膜 瓣与基质床之间会迅速形成颗粒状沉积物。治疗选择包括LASIK 角膜 瓣移除后的PTK 、DALK 和PK,按此顺序考虑。早期就诊眼科专科医生非常重要。
TGFBI基因编码细胞外基质蛋白TGFBIp(角膜上皮 素,68 kDa)。TGFBIp参与细胞粘附、迁移和增殖,并在正常角膜基质 中表达1,5,7) 。TGFBI基因发生突变后,突变型TGFBIp对蛋白水解的敏感性降低,导致其作为不溶性沉积物在角膜基质 中蓄积5,7) 。
GCD1由Arg555Trp(R555W)突变引起。突变型TGFBIp作为透明蛋白沉积在角膜基质 浅层。不含淀粉样蛋白3) 。
GCD2几乎完全由Arg124His(R124H)突变引起1,5) 。在GCD2中,透明蛋白和淀粉样蛋白均有沉积。
自噬障碍 :GCD2中已报道存在自噬障碍,导致TGFBIp降解减少,从而促进蓄积1,5) 线粒体功能障碍 :有研究表明,突变型TGFBIp本身会影响角膜 成纤维细胞,可能导致线粒体功能障碍1) 角膜新生血管 的影响角膜新生血管 的区域,沉积物有减少和再吸收的趋势。这一发现支持了沉积物集中在无血管的角膜 中央部的机制1)
LASIK 术后,TGFBIp迅速沉积在角膜 瓣和基质床之间。这被认为是由于角膜 中央部的手术操作促进了突变TGFBIp的积累1,8) 。白内障 手术时的角膜 切口(近角膜缘 )不会导致加重,因此推测与血管化的角膜缘 的距离有关1) 。Awwad等人的病理学观察表明,LASIK 术后形成的沉积物与角膜 瓣-基质床界面伤口愈合反应相关的角膜 细胞活化以及TGFBIp的聚集有关8) 。
在GCD1中,沉积物在光学显微镜下表现为均质的嗜酸性物质,Masson三色染色呈红色。在电子显微镜水平,它们被识别为直径100-500nm的棒状或梯形高电子密度结构6) 。
在GCD2中,除了透明沉积物外,还观察到淀粉样纤维(直径8-10nm)。淀粉样纤维经刚果红染色呈橙红色,在偏振光显微镜下呈现苹果绿色双折射6) 。双重染色特性有助于GCD2的病理确诊。
根据Poulsen等人的蛋白质组学分析,GCD2患者角膜 中的突变R124H-TGFBIp与正常TGFBIp相比,不易被蛋白水解酶切割,并且特定的C末端片段选择性积累6) 。这种切割抵抗性被认为是透明和淀粉样纤维形成的基础。
针对GCD2的新疗法正在开发中,但均处于研究阶段1,5) 。
氯化锂已被报道可减少TGFBI蛋白的产生。褪黑素和雷帕霉素的联合治疗可能抑制TGFBI蛋白表达,同时激活自噬以促进突变TGFBIp的降解1,5) 。
使用小干扰RNA(siRNA)或短发夹RNA(shRNA)沉默突变TGFBI表达的研究正在进行临床前研究。CRISPR/Cas9基因组编辑技术也是一个候选,但对正常等位基因或其他基因的意外脱靶效应仍是一个挑战1,5) 。
角膜 电解术角膜移植术 后复发病例的试验性使用。长期结果未知。机器学习辅助诊断 :已有报道开发出从前段照片自动识别GCD的AI模型。分子伴侣疗法 :关于化学分子伴侣(如4-苯基丁酸)辅助突变TGFBIp正确折叠的基础研究正在进行中3) 。iPS细胞来源角膜上皮 片 :使用患者来源iPS细胞制备的角膜上皮 片进行移植治疗作为未来选择正在研究中。
在日本,以日本眼科学会和日本角膜 学会为中心,关于建立TGFBI相关角膜营养不良 国家登记处的讨论仍在继续。自2020年基因检测纳入保险以来,基因诊断病例增加,日本人特有的突变模式和表型的长期随访数据正在积累3,11) 。未来,预计将建立从携带者筛查到屈光 手术适应症判断的循证诊疗体系。
对于GCD患者,以下生活指导很重要。
紫外线防护 :外出时佩戴具有防紫外线功能的太阳镜或眼镜,减少角膜 的紫外线暴露。避免眼外伤 :撞击或异物造成的角膜 损伤可能诱发复发性上皮脱落。运动或工作时使用防护眼镜。定期检查 :即使是杂合子也应每年进行一次裂隙灯 检查和视力检查 。家族检查 :建议一级亲属进行家族内筛查。隐形眼镜 :原则上可以使用软性隐形眼镜,但应缩短佩戴时间并严格执行定期更换。避免屈光 手术 :LASIK 类手术绝对禁忌。选择眼镜或隐形眼镜矫正。
Chang MS , Jun I, Kim EK. Mini-Review: Clinical Features and Management of Granular Corneal Dystrophy Type 2. Korean journal of ophthalmology : KJO. 2023;37(4):340-347. doi:10.3341/kjo.2023.0032. PMID:37336511; PMCI D:PMC10427907.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Lisch W, Weiss JS. Clinical and genetic update of corneal dystrophies. Experimental eye research. 2019;186:107715. doi:10.1016/j.exer.2019.107715. PMID:31301286.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern®. Ophthalmology. 2024;131(4):P1-P79.
Poulsen ET, Dyrlund TF, Runager K, et al. Proteomics of granular corneal dystrophy type 2 reveals altered TGFBIp proteolytic processing. J Proteome Res. 2014;13(10):4659-4667.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Awwad ST, Di Pascuale MA, Hogan RN, Forstot SL, McCulley JP, Cavanagh HD. Avellino corneal dystrophy worsening after laser in situ keratomileusis: further clinicopathologic observations and proposed pathogenesis. American journal of ophthalmology. 2008;145(4):656-61. doi:10.1016/j.ajo.2007.12.008. PMID:18243154; PMCI D:PMC2661203.
Jun RM, Tchah H, Kim TI, et al. Avellino corneal dystrophy after LASIK . Ophthalmology. 2004;111(3):463-468.
Kitazawa K, Kawasaki S, Shinomiya K, et al. Safety of anterior lamellar keratoplasty for recurrent TGFBI corneal dystrophy. Br J Ophthalmol. 2016;100(4):448-454.
日本眼科学会. 角膜 ジストロフィ遺伝子検査の保険収載について. 日本眼科学会雑誌. 2020;124:厚生労働省告示に基づく2020年4月1日収載.
复制全文后,可以粘贴到你常用的 AI 助手中提问。
打开下面的 AI 助手,并把复制的内容粘贴到聊天框。