La distrofia corneal granular (DCG) es una enfermedad corneal hereditaria caracterizada por depósitos granulares en el estroma corneal. En la segunda edición de la Clasificación Internacional de Distrofias Corneales (IC3D 2015), se clasifica como una distrofia relacionada con TGFBI epitelial-estromal 2).
Es causada por mutaciones puntuales en el gen TGFBI (cromosoma 5q31) y sigue un patrón de herencia autosómico dominante. Según la mutación, se clasifica en los siguientes dos tipos.
Clasificación
Mutación principal
Nombre alternativo / nombre antiguo
Depósitos principales
DCG1
Arg555Trp (R555W)
Granular clásica / Groenouw tipo 1
Solo hialina
GCD2
Arg124His (R124H)
Distrofia corneal de Avellino
Hialina + amiloide
GCD2 fue reportada en 1988 como un subtipo independiente de GCD1, y se denominó distrofia corneal de Avellino porque la primera familia se originó en la región de Avellino, Italia 1). Posteriormente, en 1997, se identificó el gen causante TGFBI y se encontró la mutación R124H 3). Desde la segunda edición de la clasificación IC3D (2015), se denomina oficialmente distrofia corneal granular tipo 2 (GCD2), y “Avellino” se menciona como nombre histórico 2).
La distrofia corneal granular se clasifica internacionalmente dentro del grupo de distrofias epitelio-estromales. Este grupo incluye seis enfermedades relacionadas con TGFBI: granular (GCD1, GCD2), reticular (LCD1, LCD3A), Reis-Bücklers y Thiel-Behnke, todas causadas por diferentes mutaciones puntuales en el gen TGFBI en 5q31 2,4). En la práctica oftalmológica japonesa, estas seis enfermedades se denominan colectivamente “distrofias corneales relacionadas con TGFBI”.
La distrofia corneal granular fue reportada por primera vez por Groenouw en 1890, y en ese entonces se llamaba simplemente “tipo 1 de Groenouw”. En 1938 se diferenció claramente de la distrofia reticular, y durante mucho tiempo se trató como una sola entidad. En 1988, se reportó una familia de la región de Avellino, Italia, con características tanto de distrofia granular como reticular, que luego se separó como GCD2 (tipo Avellino) 1,2). Con la revisión de la clasificación IC3D en 2015 se estableció el sistema actual, y la nomenclatura basada en el genotipo se convirtió en el estándar internacional 2).
Patrón de herencia: Autosómico dominante con alta penetrancia
Tipo 1: Frecuente en Europa y Estados Unidos; raro en Japón
Tipo 2: Abrumadoramente frecuente en Asia oriental, como Japón y Corea del Sur. La prevalencia en Corea del Sur es de aproximadamente 11.5 por cada 10,000 personas 1)
Proporción entre las distrofias corneales relacionadas con TGFBI: GCD2 representa el 72–91% en Corea del Sur y Japón, el 36% en Estados Unidos y el 3% en Polonia 1)
Datos de diagnóstico genético en Japón: En la Universidad de Yamaguchi, 234 pacientes con distrofia corneal fueron diagnosticados genéticamente durante 21 años (2000–2021), y las cuatro distrofias corneales principales (granular tipo I y II, reticular tipo I y IIIA, gelatinosa en gotas y macular) representaron aproximadamente el 96% de todos los casos. - Características en Asia Oriental: La distrofia corneal granular es abrumadoramente tipo 2 (R124H) en Asia Oriental. - Edad de inicio: Los heterocigotos de GCD2 presentan microopacidades observables solo con lámpara de hendidura desde la edad escolar, pero sin síntomas subjetivos. La disminución visual subjetiva aparece típicamente en los 40–50 años.
Diferencia de sexo: Herencia autosómica dominante, sin diferencia de sexo.
Q¿Qué significa «granular»?
A
Se refiere a una condición en la que se forman muchos pequeños grumos blancos a blanco grisáceos, bien delimitados (depósitos granulares), en el estroma superficial de la córnea central. Observados directamente con un microscopio de lámpara de hendidura, se describen como migas de pan, copos de nieve o confites. Los depósitos se originan de la proteína TGFBI mutante.
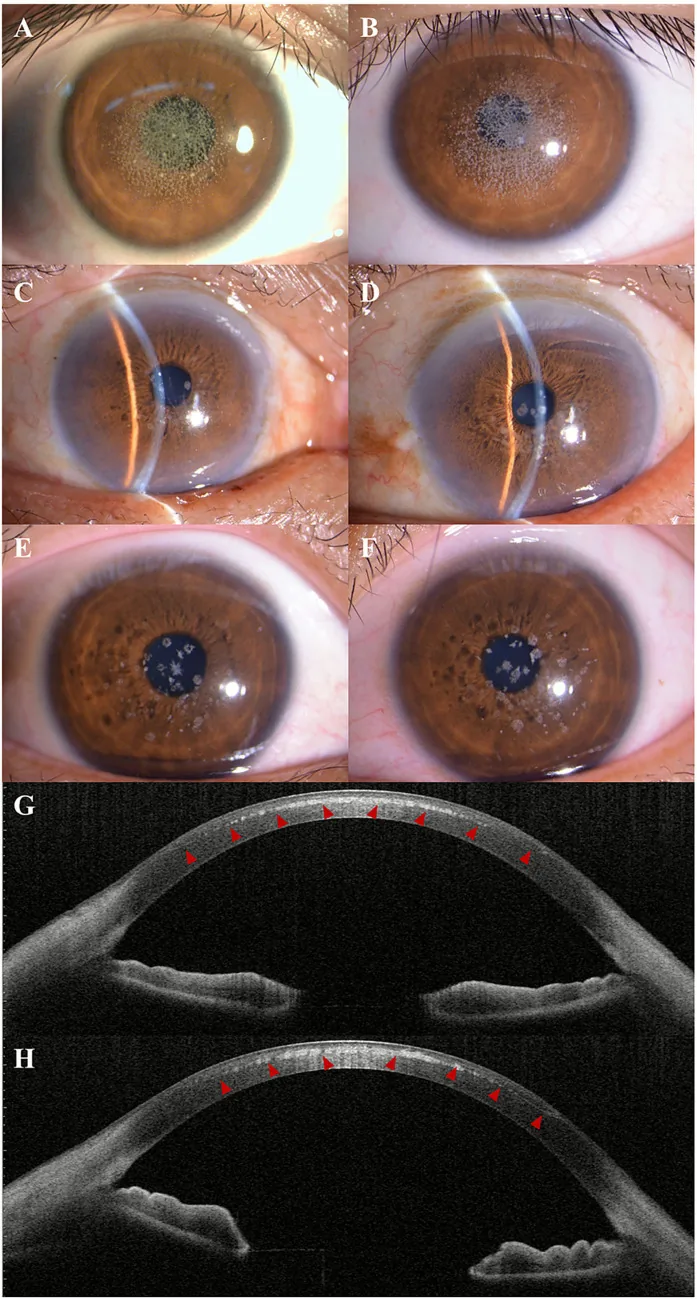
Kuang L, et al. Case Report: Post-LASIK exacerbation of granular corneal dystrophy type 2: a familial case with TGFBI mutation. Front Med (Lausanne). 2025. Figure 1. PMCID: PMC12722859. License: CC BY.
La fotografía con lámpara de hendidura muestra opacidades granulares blanco grisáceas dispersas y agrupadas en la córnea central a paracentral. La AS-OCT muestra depósitos hiperreflectivos en el estroma corneal anterior, indicando hallazgos clínicos de distrofia corneal granular.
Asintomático a leve: En heterocigotos, las etapas tempranas a intermedias a menudo no presentan disminución visual subjetiva, y muchos casos se descubren incidentalmente durante chequeos. La mayoría se queja de disminución visual en los 40–50 años.
Deslumbramiento y fotofobia: Cuando las opacidades afectan el área pupilar, los pacientes se quejan de deslumbramiento diurno y reducción de la sensibilidad al contraste.
Erosión corneal epitelial recurrente: Los depósitos dañan la capa de Bowman y la membrana basal epitelial, causando dolor ocular agudo, lagrimeo y enrojecimiento durante el sueño o al despertar.
Disminución visual: Cuando las áreas transparentes entre los depósitos se vuelven opacas, la visión disminuye progresivamente3).
Reducción de la sensibilidad al contraste: La sensibilidad al contraste a menudo disminuye antes que la agudeza visual (prueba del anillo de Landolt).
Tendencia a ceguera diurna: Debido a que la luz dispersa afecta fuertemente en condiciones brillantes, los pacientes se quejan de deslumbramiento al aire libre o al conducir.
Difícil de corregir con gafas o lentes de contacto: La dispersión de los depósitos no mejora con la corrección refractiva.
En homocigotos, la disminución visual marcada ocurre desde la infancia temprana (4-7 años) y se requiere tratamiento alrededor de los 10 años.
Hallazgos clínicos (hallazgos confirmados por el médico en el examen)
Opacidades granulares: Opacidades granulares pequeñas, bien delimitadas, de color blanco a blanco grisáceo, dispersas en la córnea central. Se describen como migas de pan o copos de nieve.
Profundidad: Subepitelial y en el estroma corneal superficial. No se extiende al limbo.
Depósitos: Solo hialina. Se tiñe de rojo con tricrómico de Masson. No contiene amiloide.
Progresión: El número de gránulos aumenta con la edad y los bordes se vuelven indistintos.
GCD2 (R124H)
Opacidades granulares: Inicio con opacidades más grandes, de color blanco a blanco grisáceo, bien delimitadas que en GCD1. Los fenotipos son diversos, incluyendo forma de confeti, lineal, estrellada, en bastón, etc.
Tipo mixto: También pueden observarse opacidades lineales finas en forma de red, similares a las de la distrofia corneal reticular.
Depósitos: Tanto hialina como amiloide. Positivo para tricrómico de Masson y positivo para rojo Congo, mostrando birrefringencia verde-amarilla bajo luz polarizada.
Progresión: Después de los 25-30 años, aparecen opacidades blancas densas en forma de bastón y estrelladas en el estroma medio. Los depósitos laminares difusos se vuelven prominentes en la capa superficial, lo que hace que la PTK sea una buena indicación3).
En ambos tipos, las opacidades se localizan en la córnea central y no se extienden a la periferia limbal. Generalmente son bilaterales con poca asimetría.
Las mutaciones homocigotas resultan en fenotipos marcadamente diferentes.
Homocigotos GCD1: Opacidades reticulares blancas a la misma profundidad desde el subepitelio hasta el estroma superficial, casi sin espacios. Al progresar, el iris y la cámara anterior se vuelven inobservables.
Homocigotos GCD2: Opacidades blancas redondas y sólidas aparecen en toda la córnea excepto la periferia, sin espacios. Es lo suficientemente grave como para ser reconocida a simple vista, y solo se conserva la transparencia limbal3). Los casos homocigotos son distrofias corneales refractarias que recurren en un corto período de 1 a 2 años incluso después de PTK o trasplante de córnea.
Q¿Cómo difiere el curso entre homocigotos y heterocigotos?
A
Los homocigotos se desarrollan en la primera infancia (4-7 años) y progresan rápidamente. Aparecen opacidades blancas en toda la córnea sin espacios, y se requiere PTK o trasplante de córnea alrededor de los 10 años. Incluso después de la cirugía, recurre en 1-2 años, siguiendo un curso refractario. Los heterocigotos progresan lentamente y generalmente pueden mantener una buena visión hasta los 40-50 años.
La GCD es causada por mutaciones puntuales en el gen TGFBI (cromosoma 5q31). El gen TGFBI codifica TGFBIp (queratoepitelina), una proteína de la matriz extracelular. La TGFBIp mutante tiene una susceptibilidad reducida a la proteólisis y se acumula como depósitos insolubles anormales en el estroma corneal1,5,7).
Las distrofias corneales relacionadas con TGFBI incluyen las siguientes2,4):
Distrofia corneal granular tipo 1 (R555W)
Distrofia corneal granular tipo 2 (R124H, anteriormente Avellino)
Antecedentes familiares: Herencia autosómica dominante; el 50% de los hijos de personas afectadas tienen riesgo de desarrollar la enfermedad.
Homocigosis: Los homocigotos para la misma mutación presentan un fenotipo más grave.
Cirugía corneal: La GCD2 puede progresar rápidamente después de un traumatismo corneal. Las opacidades empeoran significativamente especialmente después de la cirugía refractiva con láser 1,8,9).
Etnia: La GCD2 es más común en Asia Oriental (Corea, Japón). Se ha sugerido un efecto fundador 3).
Factores ambientales desconocidos: Los efectos directos de la exposición a rayos UV o la diabetes no se han establecido en la actualidad.
La GCD es un trastorno autosómico dominante con alta penetrancia. Si se diagnostica a un probando, el 50% de los familiares de primer grado (padres, hermanos, hijos) pueden portar la misma mutación. La identificación temprana de portadores asintomáticos en la familia puede ayudar a evitar futuras cirugías refractivas y establecer un seguimiento regular para monitorizar la progresión 1,5). Especialmente en jóvenes que consideran LASIK, se recomienda encarecidamente una historia familiar detallada y pruebas genéticas cuando esté indicado.
El diagnóstico clínico se basa en la observación con lámpara de hendidura de opacidades granulares bien delimitadas en el estroma anterior y en una historia familiar positiva. Se debe sospechar distrofia corneal cuando se presentan opacidades corneales (depósitos) bilaterales sin inyección ni edema corneal3).
En el diagnóstico diferencial de las distrofias corneales, primero se determina si los depósitos son “bien delimitados” o “difusos” 3). Para los depósitos granulares bien delimitados, se diferencia entre GCD1 (pequeños) y GCD2 (grandes) según el tamaño del depósito. En GCD2, la dispersión escleral revela opacidades laminares difusas entre los depósitos granulares; estas opacidades laminares son una buena indicación para PTK3).
Microscopía con lámpara de hendidura: Observación directa de opacidades granulares blancas bien delimitadas. También se utilizan dispersión escleral, retroiluminación y transiluminación.
Microscopía confocal: Revela opacidades irregulares e hiperreflectivas en forma de migas de pan entre el epitelio y la capa de Bowman.
Microscopía ultrasónica biomicroscópica (UBM): Detecta gránulos hiperreflectivos en el estroma superficial.
Topografía corneal: Proporciona información adicional sobre la densidad de las opacidades.
Prueba genética: El análisis del gen TGFBI es útil para el diagnóstico definitivo. En Japón, está cubierto por el seguro como prueba genética de distrofia corneal desde abril de 2020 3).
Fotografía del segmento anterior: Para el seguimiento a largo plazo, es importante tomar fotografías de alta calidad del segmento anterior en la visita inicial y en cada seguimiento, y conservarlas en el historial médico.
GCD1: Depósitos hialinos que se tiñen de rojo con la tinción de Masson. No contiene amiloide. La microscopía electrónica muestra depósitos en forma de bastón o trapezoidales.
GCD2: Se depositan tanto hialina (positiva a la tinción de Masson) como amiloide (positiva a la tinción de rojo Congo, birrefringencia verde manzana bajo luz polarizada). La microscopía electrónica revela depósitos electrodensos en forma de bastón y fibrillas de amiloide 1,7).
Distrofia corneal reticular tipo 1 (LCD1): Mutación TGFBI R124C. Opacidades lineales y en forma de rejilla por depósito de amiloide en el estroma. Frecuentemente asociada a erosión epitelial corneal recurrente 3)
Distrofia corneal macular (MCD): Mutación del gen CHST6. Autosómica recesiva. Opacificación difusa de toda la córnea
Distrofia corneal de Reis-Bücklers: Mutación TGFBI R124L. Opacidades en forma de mapa en la capa de Bowman
Distrofia corneal moteada (FCD): Mutación PIP5K3. Pequeñas manchas blancas en todo el estroma, generalmente asintomática
Q¿La prueba genética está cubierta por el seguro?
A
Desde abril de 2020, la prueba genética para distrofia corneal está cubierta por el seguro. Sin embargo, se requiere certificación del centro, por lo que las instalaciones que pueden realizarla son limitadas. Si los hallazgos clínicos son sospechosos o se considera cirugía refractiva como LASIK, es deseable un diagnóstico definitivo mediante prueba genética.
En la etapa inicial sin deterioro visual ni desprendimiento epitelial corneal recurrente, no es necesario tratamiento. Se considera intervención quirúrgica cuando el deterioro visual se extiende al área pupilar.
Lágrimas artificiales: Usar gotas de hialuronato de sodio al 0.1% o 0.3% de 4 a 6 veces al día para reducir la sequedad y la irritación
Lentes de contacto blandas terapéuticas: Para el desprendimiento epitelial recurrente, proteger la superficie ocular y promover la cicatrización. Generalmente se usan todo el día, requiriendo reemplazo regular
Gotas/pomada antibiótica: Para prevenir infección secundaria durante el desprendimiento epitelial, usar gotas de levofloxacino al 0.5% 3 a 4 veces al día y pomada de ofloxacino al acostarse
Solución salina hipertónica (colirio/ungüento de cloruro de sodio al 5%): Puede usarse como tratamiento coadyuvante para reducir el edema epitelial.
El tratamiento se selecciona según la profundidad de los depósitos. El objetivo final es retrasar el trasplante de córnea tanto como sea posible1,6).
PTK (Queratectomía Terapéutica)
Indicaciones: Opacidades del estroma anterior. Recomendado como tratamiento de primera línea1).
Ventajas: Repetible. Sin riesgo de rechazo del injerto. Las opacidades subepiteliales difusas son una buena indicación para PTK3).
Procedimiento: El láser excímero ablaciona aproximadamente 50 μm de estroma por sesión. La profundidad de ablación está limitada por el grosor corneal1,8).
Limitaciones: Cada ablación induce aproximadamente 1.5 D de hipermetropía. Hay un límite en el número de sesiones.
Homocigotos para GCD2: Recurrencia aproximadamente 18 meses después de la primera PTK, y después del segundo o tercer procedimiento, recurrencia en aproximadamente 3 meses1).
Heterocigotos para GCD2: La recurrencia después de PTK es relativamente lenta, con un promedio de 38.4 meses1).
Uso combinado de mitomicina C (MMC): No se recomienda el uso de MMC durante la PTK. La MMC induce apoptosis de los queratocitos en el estroma corneal, reduciendo las células responsables de la reabsorción y degradación de TGFBIp, lo que puede acelerar la recurrencia1).
Casos de exacerbación después de LASIK: Se puede realizar PTK, pero el efecto es mayor después de retirar el flap de LASIK1,8).
Manejo postoperatorio: Después de PTK, usar gotas oftálmicas antibacterianas (levofloxacino 0.5%) y gotas oftálmicas de corticosteroides (fluorometolona 0.1%) cuatro veces al día hasta la cicatrización epitelial, luego reducir gradualmente. La cicatrización epitelial generalmente toma de 3 a 5 días.
DALK (queratoplastia lamelar anterior profunda) en la práctica
La DALK es un procedimiento que preserva el endotelio. Utilizando la técnica de burbuja grande (big bubble), se diseca y elimina el estroma hasta justo por encima de la membrana de Descemet, y se sutura el estroma donante. Dado que no hay riesgo de rechazo endotelial, el pronóstico a largo plazo se considera mejor que el PK3). En un estudio de Kitazawa et al., la agudeza visual a los 5 años después de DALK para distrofias corneales relacionadas con TGFBI (incluyendo tipos granular y reticular) fue generalmente buena, y la tasa de supervivencia del injerto fue alta10). En Japón, se puede realizar bajo seguro médico.
Diagrama de flujo para la selección de la técnica quirúrgica
La selección del tratamiento quirúrgico para la GCD se considera en el siguiente orden1,6).
Depósitos limitados al estroma anterior (subepitelial hasta aproximadamente 150 μm) → PTK
Recurrencia después de múltiples PTK u opacidades profundas (150–400 μm) → DALK
Opacidades de espesor total, disfunción endotelial concurrente o casos en los que no es factible la DALK → PK
Casos refractarios con recurrencia repetida después de cualquier procedimiento (especialmente homocigotos GCD2) → Considerar enfoques de investigación como la terapia génica
Contraindicaciones de la cirugía refractiva con láser
La GCD es una contraindicación para LASIK, LASEK, PRK y SMILE. Después de la cirugía, la opacidad corneal empeora rápidamente, causando una pérdida severa de la visión 1,8,9). Tras LASIK, se forman numerosos depósitos granulares pequeños entre el flap y el lecho estromal. LASIK produce una exacerbación más grave y una peor agudeza visual final en comparación con PRK 1,8). Informes de casos de Corea del Sur y Japón describen muchos pacientes que eran asintomáticos antes de la cirugía pero desarrollaron opacidad corneal marcada meses o años después de LASIK, requiriendo PTK o trasplante de córnea8,9).
Q¿Qué sucede si se descubre GCD después de someterse a LASIK?
A
En casos de GCD que se desarrolla después de LASIK, se forman rápidamente depósitos granulares entre el flap y el lecho estromal. Las opciones de tratamiento incluyen PTK después de la eliminación del flap de LASIK, DALK y PK, consideradas en este orden. Es importante una consulta temprana con un oftalmólogo.
El gen TGFBI codifica la proteína de matriz extracelular TGFBIp (queratoepitelina, 68 kDa). TGFBIp participa en la adhesión, migración y proliferación celular, y se expresa en el estroma corneal normal 1,5,7). Las mutaciones en el gen TGFBI reducen la susceptibilidad de la TGFBIp mutante a la proteólisis, lo que lleva a su acumulación como depósitos insolubles en el estroma corneal5,7).
GCD1 es causada por la mutación Arg555Trp (R555W). La TGFBIp mutante se deposita como hialina en el estroma corneal superficial. No contiene amiloide 3).
GCD2 es causada casi exclusivamente por la mutación Arg124His (R124H) 1,5). En GCD2, se depositan tanto hialina como amiloide.
Alteración de la autofagia: Se ha reportado alteración de la autofagia en GCD2, lo que reduce la degradación de TGFBIp y promueve su acumulación 1,5)
Disfunción mitocondrial: Se ha sugerido que la TGFBIp mutante afecta a los fibroblastos corneales y puede causar disfunción mitocondrial 1)
Efecto de la neovascularización corneal: Los depósitos tienden a disminuir y reabsorberse en áreas con neovascularización corneal. Este hallazgo respalda el mecanismo por el cual los depósitos se concentran en la córnea central avascular1)
Después de LASIK, TGFBIp se deposita rápidamente entre el flap y el lecho estromal. Se cree que esto se debe a que la manipulación quirúrgica en la córnea central promueve la acumulación de TGFBIp mutante1,8). Dado que las incisiones corneales durante la cirugía de cataratas (cerca del limbo) no causan exacerbación, se presume que la distancia del limbo vascularizado es relevante1). Las observaciones patológicas de Awwad et al. sugieren que los depósitos formados después de LASIK implican la acumulación de TGFBIp junto con la activación de queratocitos asociada con la cicatrización de heridas en la interfaz flap-lecho estromal8).
En GCD1, los depósitos aparecen como material eosinofílico homogéneo bajo microscopía óptica y se tiñen de rojo con la tinción tricrómica de Masson. A nivel de microscopía electrónica, se reconocen como estructuras electrodensas en forma de bastón o trapezoidales con un diámetro de 100–500 nm6).
En GCD2, además de los depósitos hialinos, se observan fibras amiloides (8–10 nm de diámetro). Las fibras amiloides se tiñen de naranja-rojo con rojo Congo y muestran birrefringencia verde manzana bajo microscopía de luz polarizada6). Las características de tinción dual son útiles para el diagnóstico patológico definitivo de GCD2.
Según el análisis proteómico de Poulsen et al., el TGFBIp mutante R124H en córneas de pacientes con GCD2 es menos susceptible a la escisión por enzimas proteolíticas en comparación con el TGFBIp normal, y fragmentos específicos del extremo C se acumulan selectivamente6). Se sugiere que esta resistencia a la escisión es la base para la formación de fibras hialinas y amiloides.
7. Investigación más reciente y perspectivas futuras
Se ha informado que el cloruro de litio reduce la producción de proteína TGFBI. La terapia combinada con melatonina y rapamicina puede inhibir la expresión de la proteína TGFBI mientras activa la autofagia para promover la degradación de TGFBIp mutante1,5).
El silenciamiento de la expresión de TGFBI mutante mediante ARN pequeño de interferencia (siARN) o ARN en horquilla corta (shARN) se está estudiando en etapa preclínica. La tecnología de edición genómica CRISPR/Cas9 también es un candidato, pero los efectos fuera del objetivo no deseados sobre alelos normales u otros genes siguen siendo un desafío1,5).
Electrólisis corneal: Se ha informado el uso experimental en casos recurrentes después del trasplante de córnea. Los resultados a largo plazo son desconocidos.
Diagnóstico asistido por aprendizaje automático: Se ha informado el desarrollo de un modelo de IA que identifica automáticamente la GCD a partir de fotografías del segmento anterior.
Terapia con chaperonas: Está en curso la investigación básica sobre chaperonas químicas (como el ácido 4-fenilbutírico) que ayudan al plegamiento correcto de la TGFBIp mutante3).
Lámina epitelial corneal derivada de células iPS: Se está estudiando como opción futura el trasplante de láminas epiteliales corneales generadas a partir de células iPS del paciente.
En Japón, continúan los debates sobre el establecimiento de un registro nacional de distrofias corneales relacionadas con TGFBI, liderados por la Sociedad Japonesa de Oftalmología y la Sociedad Japonesa de Córnea. Desde que las pruebas genéticas fueron cubiertas por el seguro en 2020, el número de casos diagnosticados genéticamente ha aumentado, y se están acumulando datos de seguimiento a largo plazo sobre patrones de mutación y fenotipos específicos de la población japonesa3,11). En el futuro, se espera el desarrollo de sistemas clínicos basados en evidencia, desde el cribado de portadores hasta la determinación de indicaciones para cirugía refractiva.
Para los pacientes con GCD, las siguientes pautas de estilo de vida son importantes.
Protección UV: Use gafas de sol o anteojos con protección UV al salir para reducir la exposición de la córnea a los rayos UV.
Evitar traumatismos oculares: Las lesiones corneales por golpes o cuerpos extraños pueden desencadenar erosiones epiteliales recurrentes. Use gafas protectoras durante deportes o trabajo.
Examen familiar: Recomiende el cribado familiar a los familiares de primer grado.
Lentes de contacto: Los lentes blandos son generalmente posibles, pero el tiempo de uso debe ser corto y se debe seguir estrictamente el reemplazo regular.
Evitar cirugía refractiva: Las cirugías tipo LASIK están absolutamente contraindicadas. Elija corrección con gafas o lentes de contacto.
Chang MS, Jun I, Kim EK. Mini-Review: Clinical Features and Management of Granular Corneal Dystrophy Type 2. Korean journal of ophthalmology : KJO. 2023;37(4):340-347. doi:10.3341/kjo.2023.0032. PMID:37336511; PMCID:PMC10427907.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Lisch W, Weiss JS. Clinical and genetic update of corneal dystrophies. Experimental eye research. 2019;186:107715. doi:10.1016/j.exer.2019.107715. PMID:31301286.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern®. Ophthalmology. 2024;131(4):P1-P79.
Poulsen ET, Dyrlund TF, Runager K, et al. Proteomics of granular corneal dystrophy type 2 reveals altered TGFBIp proteolytic processing. J Proteome Res. 2014;13(10):4659-4667.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Awwad ST, Di Pascuale MA, Hogan RN, Forstot SL, McCulley JP, Cavanagh HD. Avellino corneal dystrophy worsening after laser in situ keratomileusis: further clinicopathologic observations and proposed pathogenesis. American journal of ophthalmology. 2008;145(4):656-61. doi:10.1016/j.ajo.2007.12.008. PMID:18243154; PMCID:PMC2661203.
Jun RM, Tchah H, Kim TI, et al. Avellino corneal dystrophy after LASIK. Ophthalmology. 2004;111(3):463-468.
Kitazawa K, Kawasaki S, Shinomiya K, et al. Safety of anterior lamellar keratoplasty for recurrent TGFBI corneal dystrophy. Br J Ophthalmol. 2016;100(4):448-454.