La distrofia corneal de Schnyder (DCS) es una distrofia corneal hereditaria causada por mutaciones en el gen UBIAD1 (UbiA prenyltransferase domain containing 1), que provoca un metabolismo lipídico anormal en la córnea y el depósito anormal de colesterol y fosfolípidos en el estroma corneal. El patrón de herencia es autosómico dominante y el locus génico se encuentra en 1p36.
Fue reportada por primera vez por Van Went y Winbaut en 1924. Después de que el oftalmólogo suizo Schnyder reportara un caso familiar de tres generaciones en 1929, el nombre de la enfermedad se estableció. En la clasificación IC3D, se clasifica como una distrofia corneal estromal.
Esta enfermedad es bilateral, con opacidad corneal que comienza en la infancia pero progresa lentamente. Una característica clínica es que la pérdida de agudeza visual es leve en relación con el grado de opacidad corneal.
Q¿Cuánto afecta la visión?
A
A pesar de los hallazgos de la lámpara de hendidura, a menudo no se observa una pérdida significativa de la agudeza visual. Sin embargo, la opacidad progresa con la edad y pueden ocurrir disfunciones visuales debido al deslumbramiento y la dispersión de la luz. Informes indican que aproximadamente el 54% de los pacientes mayores de 50 años y el 77% mayores de 70 años eventualmente requieren trasplante de córnea.
Gimenez JB, Izdebska J, Szaflik JP. Schnyder Corneal Dystrophy in an Adolescent: A Case Report With Multimodal Imaging. Cureus. 2025 Aug 11; 17(8):e89786. Figure 3. PMCID: PMC12421702. License: CC BY.
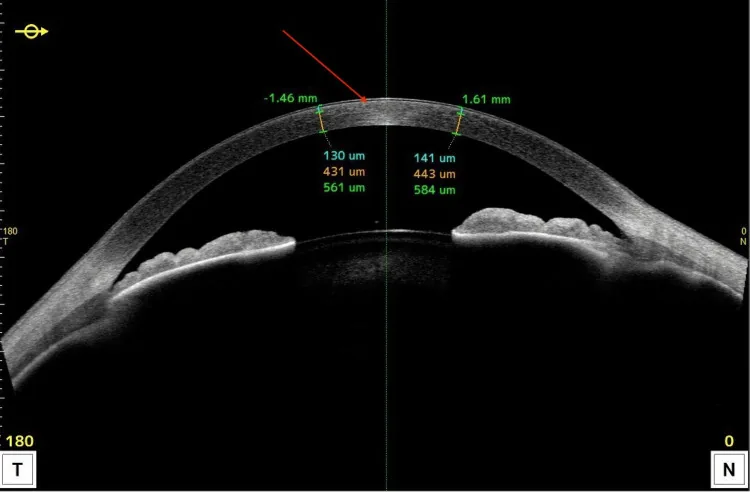
La OCT de segmento anterior muestra lesiones hiperreflectivas localizadas en la córnea anterior y cambios en el grosor. Es útil para explicar en sección transversal en qué capa se distribuyen principalmente los depósitos lipídicos.
La opacidad central y limbal progresa a todo el espesor
Cristales corneales: Se observan en aproximadamente el 50% de los casos. Se observan como una acumulación de cristales finos en forma de aguja en el estroma anterior (cerca de la membrana de Bowman).
Opacidad corneal central: Inicialmente aparece como una opacidad gris redonda u ovalada en el estroma superficial. Al progresar, se expande a las capas media y profunda.
Opacidad limbal en forma de arco senil (arcus lipoides): Aparece en la córnea periférica a partir de los 20 años. Es característico que ocurra a una edad temprana, a diferencia del arco senil típico.
Zona intermedia clara: Después de los 40 años, el área periférica media entre la opacidad central y la opacidad limbal permanece relativamente clara.
Disminución de la sensibilidad corneal: Se observa pérdida de los nervios corneales, pero no hay informes de queratopatía neurotrófica evidente.
Hallazgos patológicos: La tinción con Oil Red O muestra gotas de grasa teñidas de rojo. La microscopía electrónica revela formación de vacuolas en el estroma corneal.
La causa de la SCD es una mutación en el gen UBIAD1 (1p36). UBIAD1 codifica una preniltransferasa que sintetiza menaquinona-4 (MK-4, vitamina K2). Esta mutación genética provoca un metabolismo lipídico anormal en la córnea, lo que resulta en depósitos de colesterol en el estroma corneal. Las pruebas genéticas son útiles para el diagnóstico definitivo.
Hiperlipidemia (hipercolesterolemia): Puede asociarse con anomalías sistémicas del metabolismo lipídico.
Genu valgum (rodillas valgas): Se conoce su asociación con anomalías esqueléticas.
Malformaciones de los dedos: Raramente se asocia con malformaciones vertebrales o de los dedos.
Q¿Hay complicaciones sistémicas?
A
Se conoce la asociación con hipercolesterolemia, por lo que se recomienda la evaluación del perfil lipídico. Además, puede presentarse genu valgum y, raramente, malformaciones de los dedos o la columna vertebral.
A pesar de los hallazgos en la lámpara de hendidura, la pérdida de agudeza visual suele ser leve y muchos casos no requieren tratamiento activo. El seguimiento regular es la base.
PTK (Queratectomía Terapéutica con Láser Excímer): Se realiza para eliminar los cristales cuando los cristales subepiteliales afectan la visión.
Queratoplastia Lamelar Anterior Profunda (DALK): Se considera en casos avanzados donde la opacidad se extiende al estroma profundo.
Queratoplastia Penetrante (PKP): Se realiza cuando la opacidad severa afecta todo el espesor. Hay informes de que aproximadamente el 54% de los pacientes mayores de 50 años y alrededor del 77% mayores de 70 años finalmente requieren PKP.
Actualmente no existe farmacoterapia para detener la progresión.
Q¿Ocurre recurrencia después del trasplante de córnea?
A
Puede ocurrir recurrencia de depósitos de colesterol en el injerto después de la queratoplastia penetrante. El momento y la extensión de la recurrencia varían entre individuos, pero es necesario un seguimiento regular después del trasplante.
La SCD es causada por mutaciones en el gen UBIAD1. UBIAD1 codifica una preniltransferasa que sintetiza menaquinona-4 (vitamina K2), y la disfunción de esta enzima conduce a una alteración del metabolismo lipídico en la córnea.
Normalmente, el estroma corneal contiene cantidades muy pequeñas de colesterol y fosfolípidos. Sin embargo, las mutaciones de UBIAD1 alteran el metabolismo del colesterol HDL en particular, causando una acumulación excesiva de colesterol y fosfolípidos en el estroma corneal. Los lípidos acumulados precipitan como cristales, deteriorando la transparencia corneal.
La microscopía confocal ha confirmado la desaparición de los nervios corneales. Sin embargo, a pesar de la pérdida nerviosa, no se ha reportado queratopatía neurotrófica evidente. El mecanismo por el cual el depósito de lípidos afecta los nervios corneales no se comprende completamente.