Die Schnyder-Hornhautdystrophie (Schnyder corneal dystrophy; SCD) ist eine erbliche Hornhautdystrophie, die durch eine Mutation im UBIAD1-Gen (UbiA prenyltransferase domain containing 1) verursacht wird, was zu einer Störung des Lipidstoffwechsels in der Hornhaut und einer abnormen Ablagerung von Cholesterin und Phospholipiden im Hornhautstroma führt. Der Erbgang ist autosomal-dominant, und der Genort liegt auf 1p36.
Erstmals berichtet wurde sie 1924 von Van Went und Winbaut. Nachdem der Schweizer Augenarzt Schnyder 1929 über familiäre Fälle über drei Generationen berichtete, setzte sich der Krankheitsname durch. In der IC3D-Klassifikation wird sie zu den stromalen Hornhautdystrophien gezählt.
Die Erkrankung beginnt beidseitig, mit Hornhauttrübung bereits im Kindesalter, schreitet jedoch langsam fort. Klinisch charakteristisch ist, dass die Sehverschlechterung im Vergleich zum Ausmaß der Hornhauttrübung gering ist.
QWie stark ist die Sehbeeinträchtigung?
A
Im Vergleich zu den Spaltlampenbefunden wird oft keine signifikante Sehverschlechterung festgestellt. Mit zunehmendem Alter schreitet die Trübung jedoch fort und kann zu Blendung (Glare) und Sehstörungen durch Lichtstreuung führen. Berichten zufolge benötigen etwa 54 % der über 50-Jährigen und etwa 77 % der über 70-Jährigen letztendlich eine Hornhauttransplantation.
Gimenez JB, Izdebska J, Szaflik JP. Schnyder Corneal Dystrophy in an Adolescent: A Case Report With Multimodal Imaging. Cureus. 2025 Aug 11; 17(8):e89786. Figure 3. PMCID: PMC12421702. License: CC BY.
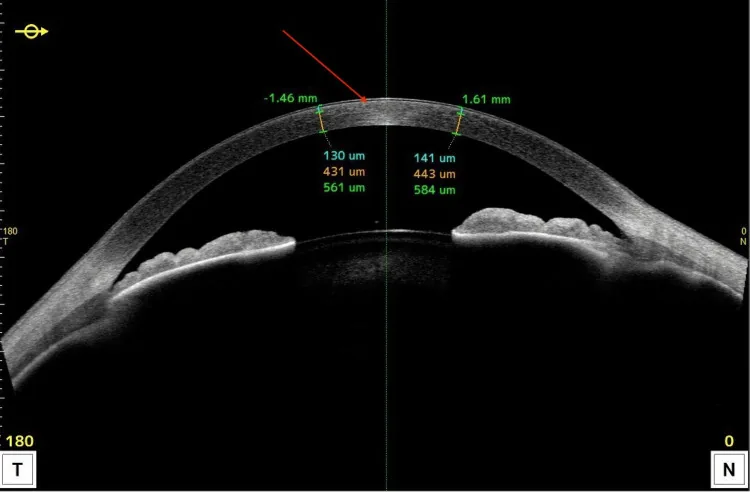
Das Vorderabschnitts-OCT zeigt hyperreflektive Läsionen, die auf den vorderen Teil der Hornhaut beschränkt sind, sowie Dickenveränderungen. Es lässt sich leicht im Querschnitt erklären, in welcher Schicht die Lipidablagerungen hauptsächlich verteilt sind.
Blendung (Glare) : verursacht durch Lichtstreuung an Kristallen oder Trübungen in der Hornhaut. Neigt dazu, mit dem Alter zuzunehmen.
Verminderung des Sehvermögens bei Helligkeit (photopisches Sehen) : Aufgrund der Lichtstreuung ist das Sehvermögen besonders bei hellem Licht vermindert.
Verminderung des Sehvermögens : Sie schreitet mit der Zunahme der Trübung langsam voran, ist aber im Verhältnis zu den Befunden oft mild.
Klinische Befunde (vom Arzt bei der Untersuchung festgestellte Befunde)
Die Hornhautbefunde entwickeln sich mit dem Alter in einem charakteristischen Muster.
Altersgruppe
Wichtigste Hornhautbefunde
Kindheit bis Jugend
Kristalline Trübung der zentralen Hornhaut
20–30 Jahre
Auftreten einer Arcus-senilis-ähnlichen limbären Trübung
40 Jahre und älter
Zentrale und limbäre Trübung schreitet in alle Schichten fort
Hornhautkristalle : Treten in etwa 50 % der Fälle auf. Sie werden als Ansammlungen feiner nadelartiger Kristalle im vorderen Stroma (nahe der Bowman-Membran) beobachtet.
Zentrale Hornhauttrübung : Anfangs erscheint sie als runde bis ovale graue Trübung im oberflächlichen Stroma. Im Fortschreiten dehnt sie sich auf die mittleren und tiefen Schichten aus.
Arcus-senilis-ähnliche limbäre Trübung (Arcus lipoides) : Tritt ab dem 20. Lebensjahr in der peripheren Hornhaut auf. Charakteristisch ist ihr Auftreten in jungen Jahren im Gegensatz zum normalen Arcus senilis.
Intermediäre klare Zone: Nach dem 40. Lebensjahr bleibt der intermediäre periphere Bereich zwischen der zentralen Trübung und der limbischen Trübung relativ klar.
Verminderte Hornhautsensibilität: Es wird ein Verlust der Hornhautnerven festgestellt, aber es gibt keine Berichte über eine offensichtliche neurotrophe Keratopathie.
Pathologisch färbt die Ölrot-O-Färbung Lipidtröpfchen rot. Elektronenmikroskopisch zeigen sich Vakuolen im Hornhautstroma.
Die Ursache der SCD ist eine Mutation im UBIAD1-Gen (1p36). UBIAD1 kodiert eine Prenyltransferase, die Menachinon-4 (MK-4, Vitamin K2) synthetisiert. Diese Genmutation führt zu einer Anomalie des Lipidstoffwechsels der Hornhaut, wobei sich Cholesterin im Hornhautstroma ablagert. Für die definitive Diagnose ist ein Gentest hilfreich.
Hyperlipidämie (Hypercholesterinämie): Kann mit einer systemischen Lipidstoffwechselstörung einhergehen.
Genu valgum (X-Beine): Eine Skelettanomalie ist bekannt.
Fingerfehlbildungen: Selten begleitet von Wirbelsäulen- oder Fingerfehlbildungen.
QGibt es systemische Komplikationen?
A
Eine Assoziation mit Hypercholesterinämie ist bekannt, und die Bewertung des Lipidprofils wird empfohlen. Darüber hinaus können Genu valgum und selten Finger- oder Wirbelsäulenfehlbildungen auftreten.
Spaltlampenmikroskopie: Grundlage der Diagnose. Beurteilung von Hornhautkristallen, zentraler Trübung und Arcus-senilis-ähnlichen Veränderungen.
Konfokale Mikroskopie: Kann das Verschwinden von Hornhautnerven bestätigen. In fortgeschrittenen Stadien werden hyperreflektive Ablagerungen beobachtet.
Vorderabschnitts-OCT: Zeigt diffuse hyperreflektive Befunde im Epithel, vorderen, mittleren und hinteren Stroma.
Definitive Diagnose und systemische Untersuchungen
Gentest: Die Identifizierung einer UBIAD1-Genmutation ermöglicht die definitive Diagnose.
Blutuntersuchung: Beurteilung des Serumlipidprofils (Gesamtcholesterin, HDL, LDL, Triglyceride).
Trotz der Befunde an der Spaltlampe ist die Sehverschlechterung oft mild, und viele Fälle benötigen keine aktive Behandlung. Regelmäßige Nachkontrollen sind die Grundlage.
PTK (therapeutische Excimer-Laser-Keratektomie) : Wird durchgeführt, um subepitheliale Kristalle zu entfernen, wenn sie das Sehvermögen beeinträchtigen.
Tiefe lamelläre Keratoplastik (DALK) : Wird bei fortgeschrittenen Fällen in Betracht gezogen, bei denen die Trübung tiefe Stromaschichten betrifft.
Durchgreifende Keratoplastik (PKP) : Wird bei starker Trübung der gesamten Hornhautdicke durchgeführt. Berichten zufolge benötigen etwa 54 % der über 50-Jährigen und etwa 77 % der über 70-Jährigen letztendlich eine durchgreifende Keratoplastik.
Derzeit gibt es keine medikamentöse Therapie, um das Fortschreiten zu stoppen.
QTritt nach einer Hornhauttransplantation ein Rezidiv auf?
A
Nach einer durchgreifenden Keratoplastik kann es zu erneuten Cholesterinablagerungen im Transplantat kommen. Zeitpunkt und Ausmaß des Rezidivs variieren individuell, aber regelmäßige Nachkontrollen nach der Transplantation sind erforderlich.
6. Pathophysiologie und detaillierter Pathomechanismus
SCD wird durch eine Mutation im UBIAD1-Gen verursacht. UBIAD1 kodiert eine Prenyltransferase, die Menachinon-4 (Vitamin K2) synthetisiert; eine Funktionsstörung dieses Enzyms führt zu einer Störung des Lipidstoffwechsels in der Hornhaut.
Normalerweise enthält das Hornhautstroma nur sehr geringe Mengen an Cholesterin und Phospholipiden. Die UBIAD1-Mutation beeinträchtigt jedoch insbesondere den HDL-Cholesterin-Stoffwechsel, was zu einer übermäßigen Ansammlung von Cholesterin und Phospholipiden im Hornhautstroma führt. Die angesammelten Lipide fallen als Kristalle aus und beeinträchtigen die Transparenz der Hornhaut.
Die konfokale Mikroskopie hat das Verschwinden der Hornhautnerven bestätigt. Trotz des Nervenverlusts wurde jedoch keine offensichtliche neurotrophe Keratopathie berichtet. Der Mechanismus, wie Lipidablagerungen die Hornhautnerven beeinflussen, ist nicht vollständig geklärt.