La dystrophie cornéenne de Schnyder (DCS) est une dystrophie cornéenne héréditaire causée par une mutation du gène UBIAD1 (UbiA prenyltransferase domain containing 1), entraînant un trouble du métabolisme lipidique dans la cornée et un dépôt anormal de cholestérol et de phospholipides dans le stroma cornéen. Le mode de transmission est autosomique dominant, et le locus génétique est situé en 1p36.
Elle a été rapportée pour la première fois en 1924 par Van Went et Winbaut. Après que l’ophtalmologiste suisse Schnyder a décrit des cas familiaux sur trois générations en 1929, le nom de la maladie s’est imposé. Dans la classification IC3D, elle est classée parmi les dystrophies cornéennes stromales.
Cette maladie débute bilatéralement, avec une opacité cornéenne apparaissant dès l’enfance, mais l’évolution est lente. Une caractéristique clinique est que la baisse de l’acuité visuelle est légère par rapport au degré d’opacité cornéenne.
QQuel est l'impact sur l'acuité visuelle ?
A
Souvent, une baisse significative de l’acuité visuelle n’est pas observée par rapport aux signes à la lampe à fente. Cependant, avec l’âge, l’opacité progresse et peut entraîner une gêne visuelle due à l’éblouissement (glare) et à la diffusion de la lumière. Des études rapportent qu’environ 54 % des patients de plus de 50 ans et 77 % de ceux de plus de 70 ans finissent par nécessiter une greffe de cornée.
Gimenez JB, Izdebska J, Szaflik JP. Schnyder Corneal Dystrophy in an Adolescent: A Case Report With Multimodal Imaging. Cureus. 2025 Aug 11; 17(8):e89786. Figure 3. PMCID: PMC12421702. License: CC BY.
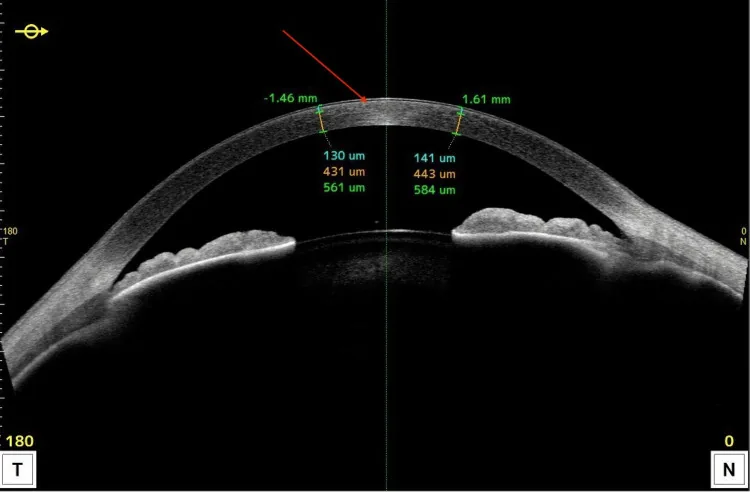
L’OCT du segment antérieur montre des lésions hyperréflectives localisées dans la partie antérieure de la cornée et des modifications d’épaisseur. Il permet d’expliquer facilement en coupe la distribution principale des dépôts lipidiques.
Les signes cornéens évoluent selon un schéma caractéristique avec l’âge.
Tranche d’âge
Principaux signes cornéens
Enfance à adolescence
Opacité cristalline centrale de la cornée
20 à 30 ans
Apparition d’une opacité limbique en forme d’arc sénile
40 ans et plus
Opacité centrale et limbique progressant sur toute l’épaisseur
Cristaux cornéens : observés dans environ 50 % des cas. Ils apparaissent sous forme d’accumulations de fins cristaux en aiguille dans le stroma antérieur (près de la membrane de Bowman).
Opacité cornéenne centrale : au début, elle se présente comme une opacité grise ronde à ovale dans le stroma superficiel. En progressant, elle s’étend aux couches moyennes et profondes.
Opacité limbique en arc sénile (arcus lipoides) : apparaît à partir de la vingtaine dans la région périphérique de la cornée. Elle se caractérise par sa survenue à un âge jeune, contrairement à l’arc sénile habituel.
Zone claire intermédiaire : après 40 ans, la zone périphérique intermédiaire entre l’opacité centrale et l’opacité limbique reste relativement transparente.
Hypoesthésie cornéenne : on observe une disparition des nerfs cornéens, mais aucun cas de kératopathie neurotrophique manifeste n’a été rapporté.
En histopathologie, la coloration Oil Red O colore les gouttelettes lipidiques en rouge. En microscopie électronique, on observe des vacuoles dans le stroma cornéen.
La cause de la SCD est une mutation du gène UBIAD1 (1p36). UBIAD1 code une prényltransférase qui synthétise la ménaquinone-4 (MK-4, vitamine K2). Cette mutation génétique entraîne une anomalie du métabolisme lipidique cornéen, avec dépôt de cholestérol dans le stroma cornéen. Le test génétique est utile pour le diagnostic définitif.
Hyperlipidémie (hypercholestérolémie) : peut être associée à une dyslipidémie systémique.
Genu valgum : une anomalie squelettique est connue.
Malformations des doigts : rarement associées à des malformations vertébrales ou digitales.
QY a-t-il des complications systémiques ?
A
L’association avec une hypercholestérolémie est connue, et l’évaluation du profil lipidique est recommandée. De plus, un genu valgum et, rarement, des malformations des doigts ou de la colonne vertébrale peuvent être présents.
Malgré les signes observés à la lampe à fente, la baisse de l’acuité visuelle est souvent légère, et de nombreux cas ne nécessitent pas de traitement actif. La surveillance régulière est la règle.
PTK (kératectomie photothérapeutique au laser excimer) : réalisée pour retirer les cristaux sous-épithéliaux cornéens lorsqu’ils affectent la vision.
Kératoplastie lamellaire profonde (DALK) : envisagée dans les cas avancés où l’opacité s’étend aux couches profondes du stroma.
Kératoplastie transfixiante (PKP) : réalisée en cas d’opacité sévère touchant toute l’épaisseur de la cornée. Des rapports indiquent qu’environ 54 % des patients de plus de 50 ans et 77 % de ceux de plus de 70 ans finissent par nécessiter une kératoplastie transfixiante.
Il n’existe actuellement aucun traitement médicamenteux pour arrêter la progression.
QY a-t-il une récidive après transplantation cornéenne ?
A
Il est possible que des dépôts de cholestérol réapparaissent sur le greffon après une kératoplastie transfixiante. Le délai et l’étendue de la récidive varient selon les individus, mais une surveillance régulière après la greffe est nécessaire.
La DCS est due à une mutation du gène UBIAD1. UBIAD1 code une prényltransférase qui synthétise la ménaquinone-4 (vitamine K2) ; un dysfonctionnement de cette enzyme entraîne une perturbation du métabolisme lipidique dans la cornée.
Normalement, la cornée contient de très faibles quantités de cholestérol et de phospholipides. Cependant, la mutation UBIAD1 altère particulièrement le métabolisme du HDL-cholestérol, entraînant une accumulation excessive de cholestérol et de phospholipides dans le stroma cornéen. Les lipides accumulés précipitent sous forme de cristaux, perturbant la transparence de la cornée.
La microscopie confocale a confirmé la disparition des nerfs cornéens. Cependant, malgré la perte nerveuse, aucune kératopathie neurotrophique évidente n’a été rapportée. Le mécanisme par lequel les dépôts lipidiques affectent les nerfs cornéens n’est pas entièrement élucidé.