Schnyder corneal dystrophy (SCD) is a hereditary corneal dystrophy caused by mutations in the UBIAD1 (UbiA prenyltransferase domain containing 1) gene, leading to abnormal lipid metabolism in the cornea and abnormal deposition of cholesterol and phospholipids in the corneal stroma. The inheritance pattern is autosomal dominant, and the gene locus is on 1p36.
It was first reported by Van Went and Winbaut in 1924. After the Swiss ophthalmologist Schnyder reported a family spanning three generations in 1929, the disease name became established. In the IC3D classification, it is classified as a stromal corneal dystrophy.
This disease is bilateral, with corneal opacification beginning in childhood but progressing slowly. A clinical feature is that visual acuity loss is mild relative to the degree of corneal opacification.
QHow much does it affect vision?
A
Despite slit-lamp findings, significant visual acuity loss is often not observed. However, opacification progresses with age, and visual dysfunction due to glare and light scattering may occur. Reports indicate that approximately 54% of patients over 50 years old and 77% over 70 years old eventually require corneal transplantation.
Gimenez JB, Izdebska J, Szaflik JP. Schnyder Corneal Dystrophy in an Adolescent: A Case Report With Multimodal Imaging. Cureus. 2025 Aug 11; 17(8):e89786. Figure 3. PMCID: PMC12421702. License: CC BY.
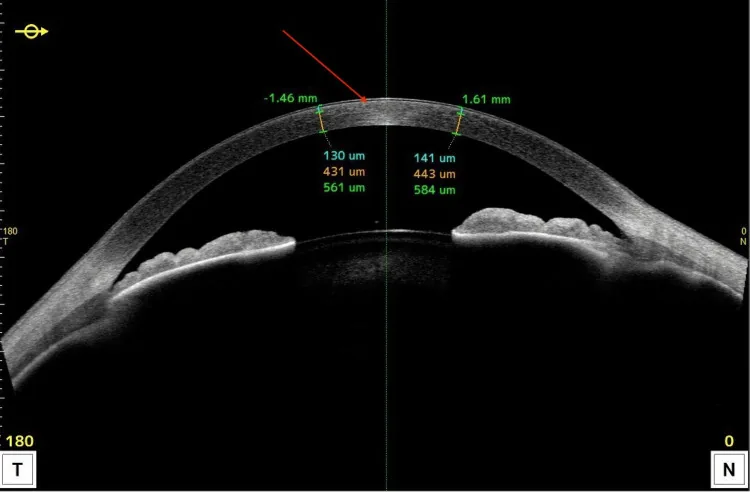
Anterior segment OCT shows hyperreflective lesions localized to the anterior cornea and changes in thickness. It is useful for explaining in cross-section which layer the lipid deposits mainly distribute.
Corneal findings progress in a characteristic pattern with age.
Age group
Main corneal findings
Childhood to teens
Crystalline opacity in the central cornea
20s to 30s
Arcus-like limbal opacity appears
40s and older
Central and limbal opacity progresses to full thickness
Corneal crystals: Observed in about 50% of cases. Seen as an accumulation of fine needle-like crystals in the anterior stroma (near Bowman’s layer).
Central corneal opacity: Initially appears as a round to oval gray opacity in the superficial stroma. As it progresses, it expands to the middle and deep layers.
Arcus lipoides (arcus senilis-like limbal opacity): Appears in the peripheral cornea from the 20s. It is characteristic that it occurs at a young age, unlike typical arcus senilis.
Clear intermediate zone: After the 40s, the mid-peripheral area between the central opacity and the limbal opacity remains relatively clear.
Decreased corneal sensitivity: Loss of corneal nerves is observed, but there are no reports of obvious neurotrophic keratopathy.
Pathological findings: Oil red O staining shows fat droplets stained red. Electron microscopy reveals vacuole formation in the corneal stroma.
The cause of SCD is a mutation in the UBIAD1 gene (1p36). UBIAD1 encodes a prenyltransferase that synthesizes menaquinone-4 (MK-4, vitamin K2). This gene mutation leads to abnormal lipid metabolism in the cornea, resulting in cholesterol deposition in the corneal stroma. Genetic testing is useful for definitive diagnosis.
Hyperlipidemia (hypercholesterolemia): May be associated with systemic lipid metabolism abnormalities.
Genu valgum (knock-knee): Known to be associated with skeletal abnormalities.
Finger deformities: Rarely associated with spinal or finger deformities.
QAre there systemic complications?
A
Hypercholesterolemia is known to be associated, and evaluation of the lipid profile is recommended. Additionally, genu valgum and rarely finger or spinal deformities may occur.
Despite slit-lamp microscope findings, visual acuity loss is often mild, and many cases do not require active treatment. Regular follow-up is the mainstay.
Penetrating Keratoplasty (PKP): Performed when severe opacity involves the full thickness. Reports indicate that approximately 54% of patients over 50 years old and about 77% over 70 eventually require PKP.
Currently, there is no drug therapy to halt progression.
QDoes recurrence occur after corneal transplantation?
A
Cholesterol deposition may recur in the graft after penetrating keratoplasty. The timing and extent of recurrence vary among individuals, but regular follow-up after transplantation is necessary.
SCD is caused by mutations in the UBIAD1 gene. UBIAD1 encodes a prenyltransferase that synthesizes menaquinone-4 (vitamin K2), and dysfunction of this enzyme leads to disruption of lipid metabolism in the cornea.
Normally, the corneal stroma contains very small amounts of cholesterol and phospholipids. However, UBIAD1 mutations impair the metabolism of HDL cholesterol in particular, causing excessive accumulation of cholesterol and phospholipids in the corneal stroma. The accumulated lipids precipitate as crystals, impairing corneal transparency.
Confocal microscopy has confirmed the disappearance of corneal nerves. However, despite nerve loss, no obvious neurotrophic keratopathy has been reported. The mechanism by which lipid deposition affects corneal nerves is not fully understood.