Lattice corneal dystrophy (LCD) is a hereditary corneal dystrophy in which amyloid deposits in the corneal stroma, producing lattice-like linear opacities. It is a historically old disease described in the 1890s, and in the IC3D (International Committee for Classification of Corneal Dystrophies) 2nd edition clinical-genetic classification, it is unified into LCD1 and its variants (formerly types 3, 3A, 1/3A, and 4)4).
LCD1, granular corneal dystrophy, Reis-Bücklers corneal dystrophy, and Thiel-Behnke corneal dystrophy form a group of diseases as “TGFBI-related dystrophies.” The causative gene TGFBI (transforming growth factor beta-induced gene) is located on the long arm of chromosome 5 (5q31) and follows an autosomal dominant inheritance pattern. TGFBI protein (TGFBIp, kerato-epithelin, βig-h3) is produced by corneal epithelial cells and distributed throughout the entire cornea. In the corneal stroma, it is involved in collagen fiber assembly. Even with mutations in the same gene, differences in mutation site and amino acid substitution lead to significant divergence in deposited material (hyaline or amyloid) and clinical presentation5).
The representative mutation of LCD1 is R124C, where arginine at position 124 of the TGFBI gene is replaced by cysteine. In the variant type LCD IIIA, mutations such as L527R have been reported.
The abnormal protein accumulated in the cornea stains red with Congo red and shows characteristic apple-green birefringence under polarized light, confirming it as amyloid. This finding has been a classic histological diagnostic indicator of amyloidosis since the 19th century6).
The type formerly called “lattice corneal dystrophy type 2” is an ocular manifestation of systemic gelsolin-type amyloidosis (GSN-AMYL, Meretoja syndrome). In the current IC3D classification, it is classified under “familial amyloidosis” and is treated independently from classic LCD4,10). This syndrome, first described by Meretoja in Finland in 1969, is a hereditary disease characterized by lattice corneal opacities along with progressive cranial neuropathy, skin laxity, and systemic symptoms10,11). Since differentiation between the two is important in clinical practice, both are described together in this article.
Double-contour filamentous opacities in the pupillary area, recurrent epithelial erosions
LCD IIIA (variant type)
TGFBI (5q31)
L527R, etc.
After 40s
Thick rope-like lattice lines in deep stroma, no epithelial involvement
GSN type (Meretoja)
GSN (9q34)
D187N, p.Glu580Lys2)
30s to 40s
Radial lattice lines in periphery, systemic amyloidosis
In Japan, the most frequent TGFBI-related dystrophy is granular type II (Avellino type, R124H), and LCD1 is less common. However, since both types diverge due to only a few base differences in the same TGFBI gene, genetic testing is recommended for confirmation in cases with overlapping clinical features. The exact prevalence of LCD overall in Japan has not been reported, but it is relatively rare among corneal dystrophies.
QWhat is the difference between LCD1 and Meretoja syndrome?
A
LCD1 is a corneal-limited amyloid deposition caused by TGFBI gene mutations, typically starting in the pupillary zone in the teens to 20s and frequently associated with recurrent epithelial erosions. In contrast, Meretoja syndrome (formerly LCD2, GSN type) is an ocular manifestation of systemic amyloidosis caused by GSN (gelsolin) gene mutations, starting in the peripheral cornea in the 30s to 40s, with central transparency preserved for a long time. Meretoja syndrome is accompanied by systemic symptoms such as skin laxity, mask-like facies, peripheral neuropathy, and cardiac arrhythmias2,10). In the IC3D second edition, Meretoja syndrome is classified independently from lattice corneal dystrophy4).
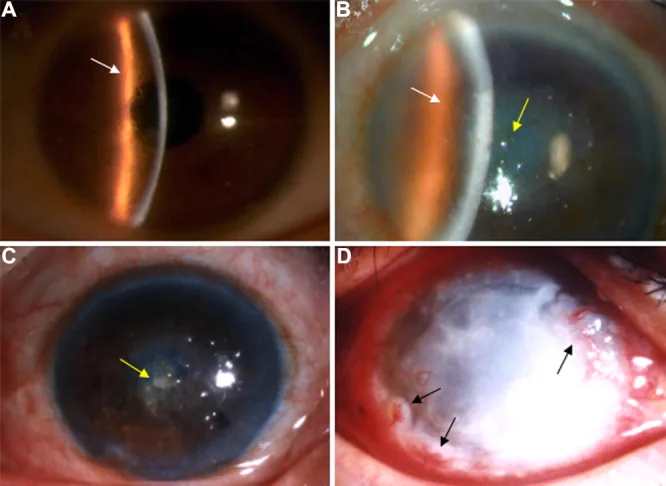
Liu Z, et al. An R124C mutation in TGFBI caused lattice corneal dystrophy type I with a variable phenotype in three Chinese families. Mol Vis. 2008. Figure 2. PMCID: PMC2443752. License: CC BY.
Slit-lamp photograph shows branching lattice lines and central-dominant opacities in the corneal stroma. This image represents typical clinical findings of lattice corneal dystrophy.
In LCD1, many patients are asymptomatic during childhood, with only fine opacities detectable by retroillumination on slit-lamp microscopy. After the teens or twenties, recurrent corneal erosions (RCE) occur repeatedly, causing acute eye pain, photophobia, tearing, and foreign body sensation upon waking. Around age 30, white opacities become apparent in the anterior stroma of the central cornea, and visual acuity progressively declines after age 40.
In LCD IIIA (variant type), epithelial damage usually does not occur, and the main complaint is gradual visual decline after age 40.
In former LCD2 (Meretoja syndrome), ocular symptoms appear in the 30s to 40s, but significant visual impairment is often delayed until the 60s 11). Systemic symptoms such as eyelid skin laxity, mask-like facies, progressive cranial neuropathy, and cardiac arrhythmias often precede or accompany the ocular findings 2,10).
Slit-lamp microscopy findings by disease type are shown below.
LCD1 (Classic Type)
Initial site: Appears as fine punctate and linear opacities in the Bowman layer to anterior stroma of the pupillary area in both eyes.
Lattice lines: Filamentous or linear opacities with double contours intertwine to form reticular or star-shaped opacities.
Advanced stage: Egg-yolk-shaped or round milky-white opacities develop in the central cornea.
Retroillumination: Thin, translucent lattice lines that are difficult to see under direct illumination become clearly visible.
Fluorescein staining: Reduced epithelial adhesion causes the surface to appear rough.
Recurrent epithelial erosion: Occurs frequently because deposits extend to the epithelial basement membrane and Bowman layer.
LCD IIIA (Variant Type)
Lattice lines: Thick, long lattice lines in the middle to deep stroma, sometimes with dendritic branching. Observable even under direct illumination.
Phenotypes: There are three patterns: (1) lattice lines only, (2) small granular deposits only, and (3) a mixture of both. In the same individual, the left and right eyes may show different phenotypes, and unilateral cases also exist.
Epithelium: Usually, no epithelial damage occurs.
Homozygotes: In L527R homozygotes, lattice lines are thicker and central granular deposits are larger, but the difference is not as pronounced as between heterozygotes and homozygotes for R124H (granular type II).
GSN type (Meretoja)
Lattice lines: A small number of lattice deposits lacking delicacy appear radially from the periphery.
Central transparency: Central transparency is maintained for a long time after onset.
Epithelial erosion: Rare.
Systemic findings: Facial changes such as mask-like facies, protruding lips with movement disorders, drooping ears, and blepharochalasis are observed2).
In LCD1, some cases show particularly strong central circular opacity. There is a report of a 56-year-old R124C heterozygote who required corneal transplantation due to central circular opacity.
QCan LCD1 be diagnosed in children?
A
Most cases of LCD1 in childhood are asymptomatic, and abnormalities are difficult to detect under direct illumination alone. Detailed observation using retroillumination or sclerotic scatter of the slit-lamp microscope can reveal fine punctate to linear opacities in the superficial central stroma. In children with recurrent corneal epithelial erosions, LCD1 should be considered, and evaluation including family history and corneal examination of both parents is recommended. TGFBI genetic testing is useful for definitive diagnosis.
The causative genes and representative mutations of lattice corneal dystrophy are summarized below.
TGFBI-related (LCD1, LCD IIIA, LCD IV)
Locus: 5q31 (TGFBI gene).
Inheritance: Autosomal dominant.
LCD1 common mutation: R124C (Arg124Cys) is the most frequent5).
LCD IIIA common mutation: L527R (Leu527Arg) and others have been reported. Homozygous cases exist.
De novo mutation: A de novo mutation of TGFBI L509P presenting with LCD IIIA phenotype has been reported1). The parents had no mutation, and the mutation was inherited by one child1).
Role of TGFBIp: Produced by corneal epithelium, distributed throughout the cornea, and involved in collagen fiber assembly in the stroma5).
GSN-related (Meretoja syndrome, formerly LCD2)
Locus: 9q34 (GSN gene, gelsolin).
Inheritance: Autosomal dominant.
Classic mutation: D187N (Finnish type) is most common; p.Asp187Tyr has also been reported10,11).
Novel mutation: p.Glu580Lys reported in a Slovenian family is located at the G4-G5 domain boundary, causing electrostatic repulsion due to substitution from negative to positive charge2).
Clinical features: In addition to corneal lattice opacities, systemic amyloidosis with skin laxity, cardiac arrhythmia, renal impairment, and optic neuropathy2).
Because it is a hereditary disease, family history is the most important risk factor. However, de novo mutations can occur in TGFBI, so absence of family history does not rule it out1). The inheritance pattern is autosomal dominant; if either parent carries the mutation, there is a 50% chance of transmission to offspring. No sex difference is observed, and racial differences are not clear for LCD1, but Meretoja syndrome is known to cluster in Finnish families11).
The contribution of environmental factors is not clear; the onset and progression of this disease are primarily determined by genotype. However, the frequency of recurrent epithelial erosions may be exacerbated by dry environments, contact lens wear, or trauma. Refractive surgeries (LASIK, SMILE, etc.) can cause rapid worsening of TGFBI-related dystrophies, and caution is needed in cases with family history during preoperative screening5).
For differentiation among LCD1, variant types, and GSN type, slit-lamp findings, histological findings, and genetic findings should be integrated.
Clinical Examination
Slit-lamp microscopy: Early lattice lines are easily overlooked under direct illumination. Use retroillumination to detect fine opacities against the pupil background, and indirect illumination to detect translucent thin lattice lines.
Fluorescein staining: In LCD1, reduced epithelial adhesion results in rough staining. It is also useful for assessing the extent of epithelial erosion.
Corneal confocal microscopy: Enables observation of deposits in the stroma at the cellular level.
Definitive Diagnosis
Genetic testing: Detection of mutations in the TGFBI gene and GSN gene confirms the disease type. Even with the same phenotype, different mutations can alter the rate of recurrence and progression, directly impacting treatment planning.
Pathological examination: Stains red with Congo red and shows apple-green birefringence under polarized light, confirming amyloid6).
Immunohistochemistry: Differentiation of disease types is possible using anti-TGFBIp antibody and anti-gelsolin antibody.
Family history taking: Since it is an autosomal dominant disorder, confirming corneal findings in parents and siblings supports the diagnosis.
Granular corneal dystrophy type II (Avellino type, TGFBI R124H): The most common TGFBI-related dystrophy in Japan, showing a mixture of granular deposits and lattice lines. Genetic testing is reliable for differentiation from LCD1.
Secondary corneal amyloidosis: Not hereditary; amyloid deposits secondarily due to chronic ocular surface irritation such as trichiasis or keratoconus. Differentiating points include absence of family history and presence of underlying disease.
Macular corneal dystrophy: Autosomal recessive inheritance due to CHST6 gene mutation, with diffuse ground-glass opacity and endothelial abnormalities.
Gelatinous drop-like corneal dystrophy: Autosomal recessive inheritance due to TACSTD2 gene mutation, presenting with milky gelatinous elevations. Relatively common in Japan.
QWhy is genetic testing important?
A
Even if lattice corneal dystrophy has similar phenotypes, differences in causative genes and mutation sites lead to significant variations in progression rate, recurrence frequency, treatment options, and presence of systemic complications. LCD1 with TGFBI mutation and Meretoja syndrome with GSN mutation have fundamentally different treatment strategies and need for systemic evaluation2,10). Furthermore, de novo mutations have been reported, making it impossible to determine the disease type based solely on family history1). Genetic testing is essential for definitive diagnosis and classification.
In childhood to young adulthood, when asymptomatic or with only fine opacities, follow-up observation is performed. Progression is assessed by slit-lamp examination every six months to one year.
Management of Recurrent Corneal Epithelial Erosions
For recurrent epithelial erosions, a core symptom of LCD1, the following conservative therapy is the first step.
Acute treatment: Continuous wear of therapeutic soft contact lenses to protect the corneal epithelium. Combined with antibacterial eye drops to prevent secondary infection. Apply eye ointment for lubrication and epithelial protection.
Recurrence prevention: Administration of eye ointment before bedtime is effective in suppressing recurrence of RCE attacks. In dry environments, use artificial tears or lubricants during the day as well.
In LCD1, where amyloid deposits are mainly in the superficial cornea, phototherapeutic keratectomy (PTK) using excimer laser is the first choice for cases with severe central opacity or recurrent corneal epithelial erosions7,8). Early recurrence usually does not occur, but recurrence over time is inevitable; PTK can be performed up to about two times on the same eye.
In heterozygotes, recurrence is slow and few cases require retreatment. In homozygotes, recurrence tends to occur earlier than in heterozygotes. The recurrence rate after PTK increases over time, similar to other TGFBI-related dystrophies, and long-term follow-up confirms some recurrence findings in many cases8).
As a case demonstrating the efficacy of PTK, in a case of LCD IIIA due to a de novo mutation in TGFBI L509P, PTK of 60 µm was performed under FD-OCT guidance, and best-corrected visual acuity (BCVA) improved from 20/400 to 20/501). No visual decline or significant recurrence was observed at 45 months postoperatively1).
According to the AAO Preferred Practice Pattern for corneal edema and opacity, PTK for granular and lattice corneal dystrophies is a “reasonable means” and may delay the transition to DALK or full-thickness corneal transplantation, but there is a risk of postoperative haze. When repeated, the use of mitomycin C is considered as a means to suppress recurrent scarring and stromal deposits, and it is warned that the risk of corneal ectasia increases when ablation exceeds the anterior one-third of the stroma or when the residual bed is less than 250 µm7).
Corneal transplantation is selected for cases with repeated recurrence or when opacity extends deeper than the mid-stroma. In LCD1, corneal transplantation is often not indicated until after age 40. In LCD, corneal endothelial cells are generally normal, so the surgical procedure is selected according to the depth of opacity.
High visual recovery but risk of rejection and recurrence
In recent years, DALK has become widely used as a new first-line treatment due to reduced risk of rejection and visual outcomes comparable to full-thickness corneal transplantation.
LCD recurrence after corneal transplantation is inevitable, with reported recurrence rates after full-thickness corneal transplantation of 17.8% at 5 years, 26% at 8 years, and 56% at 15 years 9). Recurrent opacities are usually confined to the superficial layer, so they can be removed by PTK, extending the interval until regrafting. For LCD IIIA (variant type), treatment is often not required unless visual acuity is significantly affected.
QHow effective is PTK?
A
PTK can effectively remove superficial amyloid deposits, improving visual acuity and reducing recurrent epithelial erosions. In a case of LCD IIIA, best corrected visual acuity improved from 20/400 to 20/50 after 60 µm PTK, with no recurrence for 45 months 1). Recurrence is slow in heterozygotes but early in homozygotes. Deep lesions cannot be removed by PTK, so DALK or full-thickness corneal transplantation is required for deep opacities 7).
QDoes LCD recur after corneal transplantation?
A
LCD recurrence after corneal transplantation is inevitable. Recurrence rates after full-thickness corneal transplantation are reported as 17.8% at 5 years, 26% at 8 years, and 56% at 15 years 9). However, recurrent opacities are usually confined to the superficial layer of the graft, so they can be removed by PTK, prolonging graft survival. Deep anterior lamellar keratoplasty (DALK) has a lower risk of endothelial rejection compared to full-thickness corneal transplantation and is gaining attention as a new first-line option 7).
The core pathology of LCD1 is abnormal accumulation of TGFBIp (kerato-epithelin, βig-h3). TGFBIp is normally produced by the corneal epithelium and distributed throughout the entire cornea, where it functions as a structural protein involved in collagen fiber assembly and cell adhesion in the stroma 5). The abnormal protein produced by the R124C mutation undergoes misfolding and self-aggregation, depositing as insoluble amyloid fibrils in Bowman’s layer and the superficial stroma. In advanced stages, deposits spread to the deep stroma.
Amyloid deposition alters the epithelial adhesion structures of the anterior cornea, causing degeneration of epithelial basal cells and epithelial layer degeneration accompanied by defects in Bowman’s membrane. This structural disruption forms the pathological basis of recurrent corneal epithelial erosions.
In the TGFBI gene, differences in mutation site and substituted amino acid determine the clinical phenotype. R124C causes LCD1, R124H causes granular corneal dystrophy type II (Avellino type), and R124L causes Reis-Bücklers corneal dystrophy5). The molecular mechanism by which a single amino acid difference determines the deposited material (amyloid vs. hyaline vs. both) and deposition site is not fully understood, but the domain of βig-h3 where the mutation occurs and its effect on folding stability are considered key factors.
In LCD IIIA, deep-layer dominant mutations such as L527R produce thick rope-like lattice lines and result in a late-onset type without epithelial involvement. The laminar localization of deposits can be explained by the secretion and diffusion gradient of βig-h3 from producing cells (epithelial basal cells) into the stroma, and differences in the folding stability of the mutant protein. R124C is thought to preferentially follow a pathway from folding intermediates to amyloid fibril formation, accumulating amyloid around Bowman’s layer 5). In contrast, the L527R mutation forms a relatively stable misfolded protein that slowly deposits in deeper stromal layers.
Posterior corneal amyloid and intraocular surgery risk
Conventionally, amyloid deposition in LCD1 was thought to be limited to the anterior cornea (Bowman’s layer to superficial stroma). However, recent pathological studies have shown that amyloid deposits also exist in the posterior cornea near Descemet’s membrane 3). Amyloid deposition in the posterior cornea may affect adhesion of Descemet’s membrane and contribute to its detachment during cataract surgery 3). It is suggested that a similar mechanism by which amyloid deposition impairs epithelial adhesion in the anterior cornea also operates in the posterior cornea3).
Gelsolin, the causative molecule of the former LCD2 (Meretoja syndrome), exists both in the cytoplasm and extracellularly and is a protein involved in cell motility, cell division, and apoptosis through actin binding. The classic D187N mutation, known as the Finnish type, presents with corneal lattice deposits and cranial neuropathy as the main phenotype 11). The novel p.Glu580Lys mutation reported in a Slovenian family is located at the G4-G5 domain boundary, and the substitution of negatively charged glutamic acid with positively charged lysine is thought to cause electrostatic repulsion, reducing interdomain connectivity and stability 2). Mutant gelsolin undergoes aberrant cleavage by furin and MT1-MMP in plasma, releasing 8 kDa and 5 kDa amyloid precursor fragments. These deposit in the corneal stroma, skin, blood vessel walls, peripheral nerves, and renal glomeruli, causing the multi-organ symptoms characteristic of Meretoja syndrome 2,11). Corneal deposits often precede other systemic symptoms, and ophthalmologists may be the first to diagnose this condition.
De novo mutations in the TGFBI gene causing LCD have been reported 1). Even in cases without a family history, the possibility of de novo mutations should be considered, and confirmation by genetic testing is recommended 1). The L509P mutation is rare but presents a wide range of phenotypes from Reis-Bücklers corneal dystrophy-like to LCD IIIA-like 1).
In the GSN gene, in addition to the conventional p.Asp187Asn/Tyr mutations, a novel p.Glu580Lys mutation has been reported, shown to cause systemic amyloidosis with corneal lattice dystrophy, skin laxity, cardiac arrhythmia, renal impairment, and optic neuropathy2).
Posterior Corneal Lesions and Intraocular Surgery Management
Pathological studies have shown that amyloid deposits exist in the posterior cornea of LCD1 patients and may affect adhesion of Descemet’s membrane 3). Attention should be paid to the risk of Descemet’s membrane detachment during intraocular surgeries such as cataract surgery.
This finding has clinical implications for the evaluation of cataract surgery indications and surgical planning in LCD1 patients.
More precise surgical techniques such as femtosecond laser-assisted lamellar keratectomy (FLK) and femtosecond laser-assisted lamellar keratoplasty (FALK) are being developed 12). These are being positioned as complementary options to conventional PTK, offering improved smoothness of the resection surface and highly reproducible depth control.
Since TGFBI mutations are autosomal dominant gain-of-function mutations, mutant allele-specific siRNA, antisense oligonucleotides, and allele-specific knockout using CRISPR-Cas9 are being investigated at the preclinical stage. The cornea is advantageous as a target organ for gene therapy because it allows local administration and has immune privilege. However, none have been clinically applied to date, and all require future verification of long-term safety and efficacy.
Amyloid Formation Inhibitors and Molecular Chaperones
Small molecule compounds targeting the aggregation process of TGFBIp and mutant gelsolin, molecular chaperones (such as Hsp70 inducers), and amyloid fibril binding inhibitors are being investigated at the basic research stage. For systemic gelsolin-type amyloidosis, drugs that inhibit the cleavage step of mutant gelsolin in plasma are being evaluated in some preclinical studies 2). In the future, such molecular targeted therapies are expected to become fundamental treatments replacing conventional physical resection (PTK and corneal transplantation).
Mass spectrometry-based corneal proteome analysis has suggested that in LCD1, not only TGFBIp but also multiple abnormal proteins may co-deposit in the deposits. Elucidation of the pathogenic contribution of these co-deposited proteins is underway for future clinical application.
Ji YW, Ahn H, Shin KJ, Kim TI, Seo KY, Stulting RD, et al. De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. Journal of clinical medicine. 2022;11(11). doi:10.3390/jcm11113055. PMID:35683443; PMCID:PMC9181583.
Potrc M, Volk M, de Rosa M, Pizem J, Teran N, Jaklic H, Maver A, Drnovsek-Olup B, Bollati M, Vogelnik K, Hocevar A, Gornik A, Pfeifer V, Peterlin B, Hawlina M, Fakin A. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. Int J Mol Sci. 2021;22(3):1084.
Matsumoto A, Fukuoka H, Sotozono C. Descemet’s Membrane Detachment During Cataract Surgery in Lattice Corneal Dystrophy Type I: Histopathological Analysis of Posterior Corneal Involvement. Cureus. 2025;17(3):e81431. doi:10.7759/cureus.81431. PMID:40296953; PMCID:PMC12037203.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
Lisch W, Seitz B. The clinical landmarks of corneal dystrophies. Developments in ophthalmology. 2011;48:9-23. doi:10.1159/000324075. PMID:21540629.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern. San Francisco, CA: American Academy of Ophthalmology; 2024.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea. 2003;22(1):19-21.
Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969;1(4):314-324. PMID:4313418.
Kivelä T, Tarkkanen A, Frangione B, Ghiso J, Haltia M. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja’s syndrome. Invest Ophthalmol Vis Sci. 1994;35(10):3759-3769. PMID:8088963.
Steger B, Romano V, Biddolph S, Willoughby CE, Batterbury M, Kaye SB. Femtosecond laser-assisted lamellar keratectomy for corneal opacities secondary to anterior corneal dystrophies: an interventional case series. Cornea. 2016;35(1):6-13. PMID:26509759. doi:10.1097/ICO.0000000000000665.
Copy the article text and paste it into your preferred AI assistant.
Article copied to clipboard
Open an AI assistant below and paste the copied text into the chat box.