LCD1(古典型)
初発部位:両眼の瞳孔領に Bowman 層〜実質浅層の微細点状・線状混濁として出現する。
格子状線:二重輪郭を持った糸状・線状混濁が絡み合い、網目状・星状の混濁を形成する。
進行期:中央部角膜に卵黄形あるいは円形の乳白色混濁を生じる。
反帰照明法:直接照明ではわかりにくい半透明の細い格子状線が明瞭に浮かび上がる。
フルオレセイン染色:上皮接着性の低下により「のり」が rough となる。
再発性上皮びらん:沈着物が上皮基底細胞と Bowman 膜に及ぶため高頻度に生じる。

格子状角膜ジストロフィ(lattice corneal dystrophy, LCD)は、角膜実質にアミロイドが沈着し格子状の線状混濁を生じる遺伝性角膜ジストロフィである。1890年代に記載された歴史の古い疾患であり、IC3D(International Committee for Classification of Corneal Dystrophies)第2版の臨床遺伝学的分類で LCD1 とそのvariant(従来の3型、3A型、1/3A型、4型)に統合されている4)。
LCD1、顆粒状角膜ジストロフィ、Reis-Bücklers角膜ジストロフィ、Thiel-Behnke角膜ジストロフィは、「TGFBI関連ジストロフィ」として一連の疾患群を形成する。原因遺伝子 TGFBI(transforming growth factor beta-induced gene)は第5染色体長腕(5q31)に位置し、常染色体優性遺伝の形式をとる。TGFBI蛋白(TGFBIp、kerato-epithelin、βig-h3)は角膜上皮が産生し角膜全層に分布する。角膜実質ではコラーゲン線維の構築に関与する。同一遺伝子の変異であっても、変異部位や置換アミノ酸の違いにより沈着物質(ヒアリンかアミロイドか)と臨床像が大きく分化する5)。
LCD1 の代表変異は TGFBI 遺伝子の 124 番目のアルギニンがシステインに置換される R124C である。variant 型である LCD IIIA では L527R などの変異が報告されている。
角膜に蓄積する異常蛋白は、コンゴレッド染色で赤色を呈し、偏光顕微鏡下に特有のアップルグリーン複屈折を示すことでアミロイドと確定される。この所見は 19 世紀以来の古典的なアミロイドーシスの組織診断指標である6)。
従来「格子状角膜ジストロフィ2型」と呼ばれていた型は、全身性のゲルソリン型アミロイドーシス(GSN-AMYL、Meretoja症候群)の眼症状であり、現行の IC3D 分類では「家族性アミロイドーシス」に分類され、古典的な LCD とは独立して扱われる4,10)。1969 年にフィンランドの Meretoja によって記載された本症候群は、角膜格子状混濁に加え進行性脳神経障害、皮膚弛緩、全身症状を伴う遺伝性疾患である10,11)。臨床現場では両者の鑑別が重要なため、本記事でも両者を併記する。
| 病型 | 遺伝子 | 代表変異 | 発症年齢 | 主な所見 |
|---|---|---|---|---|
| LCD1(古典型) | TGFBI(5q31) | R124C | 10〜20歳代 | 瞳孔領の二重輪郭糸状混濁、再発性上皮びらん |
| LCD IIIA(variant型) | TGFBI(5q31) | L527R 等 | 40歳代以降 | 実質深層の太いロープ状格子線、上皮障害なし |
| GSN型(Meretoja) | GSN(9q34) | D187N、p.Glu580Lys2) | 30〜40歳代 | 周辺部放射状の格子線、全身性アミロイドーシス |
本邦で頻度が高い TGFBI 関連ジストロフィは圧倒的に顆粒状 II 型(Avellino型、R124H)であり、LCD1 はこれに比べて少ない。ただし両者は同じ TGFBI 遺伝子のわずか数塩基の違いで病型が分岐するため、臨床像が重複する症例では遺伝子検査による確定が望ましい。本邦における LCD 全体の正確な有病率は報告されていないが、角膜ジストロフィ全体の中では比較的まれな部類に入る。
LCD1 は TGFBI 遺伝子変異による角膜に限局したアミロイド沈着であり、10〜20歳代に瞳孔領から発症して再発性上皮びらんを高頻度に伴う。一方、Meretoja 症候群(旧LCD2、GSN型)は GSN(ゲルソリン)遺伝子変異による全身性アミロイドーシスの眼症状で、30〜40歳代に周辺部角膜から発症し、中央部の透明性が長く保たれる。Meretoja 症候群では皮膚弛緩、仮面様顔貌、末梢神経障害、心臓不整脈などの全身症状を伴う2,10)。IC3D 第2版では Meretoja 症候群は格子状角膜ジストロフィとは独立に分類される4)。
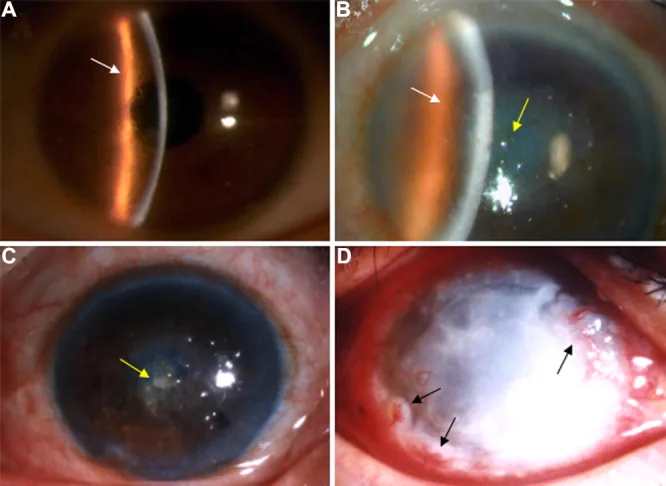
LCD1 では小児期には多くが無症状で、細隙灯顕微鏡の徹照法でようやく検出される微細混濁のみである。10〜20歳代以降に再発性角膜上皮びらん(recurrent corneal erosion, RCE)を繰り返し、起床時の急激な眼痛・羞明・流涙・異物感を反復する。30歳代頃に角膜中央部実質浅層に白色混濁が顕在化し、40歳代以降に視力低下が進行する。
LCD IIIA(variant型)では上皮障害は通常生じず、40歳以降の緩徐な視力低下が主訴となる。
旧LCD2(Meretoja症候群)では、眼症状は 30〜40歳代に出現するが、重大な視力障害は 60 歳代まで遅延することが多い11)。眼瞼皮膚弛緩、仮面様顔貌、進行性脳神経障害、心臓不整脈など全身症状が先行・並行することが多い2,10)。
病型ごとの細隙灯顕微鏡所見を以下に示す。
LCD1(古典型)
初発部位:両眼の瞳孔領に Bowman 層〜実質浅層の微細点状・線状混濁として出現する。
格子状線:二重輪郭を持った糸状・線状混濁が絡み合い、網目状・星状の混濁を形成する。
進行期:中央部角膜に卵黄形あるいは円形の乳白色混濁を生じる。
反帰照明法:直接照明ではわかりにくい半透明の細い格子状線が明瞭に浮かび上がる。
フルオレセイン染色:上皮接着性の低下により「のり」が rough となる。
再発性上皮びらん:沈着物が上皮基底細胞と Bowman 膜に及ぶため高頻度に生じる。
LCD IIIA(variant型)
格子状線:実質中層〜深層に太く長い格子状線、時に樹状分岐を呈する。直接照明でも観察可能である。
表現型:①格子状線のみ、②小粒状沈着のみ、③両者の混在、の3パターンがある。同一個体内で左右眼が異なる表現型を示す場合や片眼性症例もある。
上皮:通常、上皮障害は来さない。
ホモ接合体:L527R ホモ接合体では格子状線がより太く、中央の粒状沈着が大きいが、R124H(顆粒状II型)のヘテロ・ホモ間の差ほど顕著ではない。
GSN型(Meretoja)
格子状線:繊細さに欠ける少数の格子状沈着が、周辺部から放射状に出現する。
中央透明性:発症後長期にわたり中央部の透明性が保たれる。
上皮びらん:稀である。
全身所見:仮面様顔貌、運動障害を伴う突出した唇、垂れ下がった耳、眼瞼皮膚弛緩症などの顔貌変化を呈する2)。
LCD1 では症例によって中央部の円形混濁が特に強くなるものがあり、56 歳の R124C ヘテロ接合体で中央円形混濁から角膜移植適応となった報告がある。
小児期の LCD1 は多くが無症状で、直接照明のみでは異常が見つけにくい。細隙灯顕微鏡の徹照法や反帰照明法を用いた詳細な観察で、中央実質浅層の微細な点状〜線状の混濁を確認できる。再発性角膜上皮びらんを繰り返す小児では LCD1 を念頭に置き、家族歴の聴取と両親の角膜検査を含めた評価が推奨される。確定診断には TGFBI 遺伝子検査が有用である。
格子状角膜ジストロフィの原因遺伝子と代表変異を以下に整理する。
TGFBI関連(LCD1・LCD IIIA・LCD IV)
遺伝子座:5q31(TGFBI 遺伝子)。
遺伝形式:常染色体優性遺伝である。
LCD1 代表変異:R124C(Arg124Cys)が最頻である5)。
LCD IIIA 代表変異:L527R(Leu527Arg)などが報告されている。ホモ接合体例も存在する。
de novo変異:TGFBI L509P の de novo 変異が LCD IIIA の表現型を呈した症例が報告されている1)。両親には変異がなく、子の一人に変異が遺伝していた1)。
TGFBIpの役割:角膜上皮が産生し角膜全層に分布、実質ではコラーゲン線維構築に関与する5)。
GSN関連(Meretoja症候群、旧LCD2)
遺伝子座:9q34(GSN 遺伝子、ゲルソリン)。
遺伝形式:常染色体優性遺伝である。
古典変異:D187N(フィンランド型)が最も多く、p.Asp187Tyr も報告されている10,11)。
新規変異:スロベニア家族で報告された p.Glu580Lys は G4-G5 ドメイン境界に位置し、負電荷から正電荷への置換により静電的反発を生じさせる2)。
臨床像:角膜格子状混濁に加え、皮膚弛緩、心臓不整脈、腎障害、視神経障害を伴う全身性アミロイドーシスを呈する2)。
遺伝性疾患であるため家族歴が最も重要なリスク因子である。ただし TGFBI では de novo 変異も生じうるため、家族歴がないことだけでは除外できない1)。遺伝形式は常染色体優性であり、親のいずれかが変異保有者であれば子に 50% の確率で伝達される。性差は認められず、人種差も LCD1 では明瞭ではないが、Meretoja 症候群はフィンランドに多発家系が集積することで知られる11)。
環境因子の寄与は明らかでなく、本症の発症・進行は原則として遺伝子型によって決定される。ただし再発性上皮びらんの頻度は、乾燥環境やコンタクトレンズ装用、外傷などで増悪することがある。屈折矯正手術(LASIK、SMILE 等)は TGFBI 関連ジストロフィの急速悪化を引き起こす可能性があり、術前スクリーニングで家族歴のある症例には注意が必要である5)。
LCD1 と variant 型、さらに GSN 型の鑑別には、細隙灯所見・組織所見・遺伝子所見を総合する。
臨床検査
確定診断
遺伝子検査:TGFBI 遺伝子および GSN 遺伝子の変異検出により病型を確定する。同一表現型でも変異が異なると再発・進行の速度が変わるため、治療計画にも直結する。
病理検査:コンゴレッド染色で赤色を呈し、偏光顕微鏡下にアップルグリーンの複屈折を示すことでアミロイドと確定する6)。
免疫組織化学:抗TGFBIp 抗体、抗ゲルソリン抗体による病型鑑別が可能である。
家族歴聴取:常染色体優性遺伝のため両親・同胞の角膜所見の確認が診断を支持する。
格子状角膜ジストロフィは表現型が似ていても原因遺伝子と変異部位が異なると、進行速度・再発頻度・治療選択・全身合併症の有無が大きく変わる。TGFBI 変異のLCD1 と GSN 変異の Meretoja 症候群では治療方針と全身精査の必要性が根本的に異なる2,10)。さらに de novo 変異があり家族歴だけでは病型を決定できない症例も報告されており1)、遺伝子検査が確定診断と病型分類に不可欠である。
格子状角膜ジストロフィの治療は、IC3D 分類に準拠した以下の段階的アプローチに基づいている。
小児期〜若年期で無症状ないし微細混濁のみの段階では経過観察とする。半年〜1年ごとの細隙灯検査で進行度を評価する。
LCD1 の中核症状である再発性上皮びらんに対しては、以下の保存療法が第一段階となる。
角膜表層のアミロイド沈着が主体である LCD1 で、中央部混濁が強いもの、あるいは再発性角膜上皮びらんが繰り返し出現する場合、エキシマレーザーを用いた phototherapeutic keratectomy(PTK、治療的レーザー角膜切除術)が第一選択である7,8)。通常は早期再発は生じないが、経時的に再発は不可避であり、PTK 治療は同一眼に対して2回程度まで施行可能である。
ヘテロ接合体では再発は緩徐で再治療を要する例は少ない。ホモ接合体ではヘテロ接合体に比べ早期に再発する傾向がある。PTK 後の再発率は他の TGFBI 関連ジストロフィと同様、時間経過とともに増加し、長期観察では多くの症例で何らかの再発所見が確認される8)。
PTK の有効性を示す症例として、TGFBI L509P の de novo 変異による LCD IIIA の症例では、FD-OCT ガイド下に 60 µm の PTK を施行し、最高矯正視力(BCVA)が 20/400 から 20/50 に改善した1)。術後 45 ヶ月時点で視力低下や有意な再発は認められなかった1)。
AAO の角膜浮腫・混濁 Preferred Practice Pattern では、顆粒状および格子状角膜ジストロフィに対する PTK は「合理的選択肢(reasonable means)」であり、DALK や全層角膜移植への移行を遅延させうるが、術後ヘイズのリスクがあるとされる。繰り返し施行時にはマイトマイシンC併用が再発性瘢痕や実質沈着を抑制する手段として検討されており、アブレーションが実質前1/3を超える場合や残存ベッドが 250 µm 未満の場合には角膜拡張症のリスクが高まると警告されている7)。
再発を繰り返す例、あるいは混濁が実質中層より深い層に及ぶ場合は角膜移植を選択する。LCD1 では通常 40 歳を過ぎるまで角膜移植の適応とならないことが多い。LCD では角膜内皮細胞は原則として正常であるため、混濁の深さに応じて術式を選択する。
| 術式 | 適応 | 特徴 |
|---|---|---|
| 表層角膜移植 | 浅層の混濁 | 低侵襲、拒絶反応が少ない |
| 深層層状角膜移植(DALK) | 中層〜深層の混濁 | 内皮を温存し拒絶リスクが低い |
| 全層角膜移植(PK) | 全層に及ぶ混濁 | 視力回復力は高いが拒絶・再発リスクあり |
近年は拒絶反応のリスク低減と全層角膜移植に匹敵する視力転帰から、DALK が新たな第一選択として広く用いられるようになっている。
角膜移植後の LCD 再発は不可避の現象であり、全層角膜移植後の再発率は 5 年で 17.8%、8 年で 26%、15 年で 56% と報告されている9)。再発混濁は通常表層に限局するため、PTK で除去でき再移植までの期間を延長できる。LCD IIIA(variant型)については、視力への影響が強くならない限り治療を要しないことが多い。
LCD1 の病態の中心は TGFBIp(kerato-epithelin、βig-h3)の異常蓄積である。TGFBIp は本来、角膜上皮が産生して角膜全層に分布し、実質ではコラーゲン線維の構築と細胞接着に関与する構造蛋白である5)。R124C 変異により産生された異常蛋白はミスフォールディングと自己凝集を起こし、不溶性のアミロイド原線維として Bowman 層〜実質浅層に沈着する。進行期には沈着が実質深層へ広がる。
アミロイド沈着は前部角膜の上皮接着構造に変化を生じさせ、上皮基底細胞の変性、Bowman 膜の欠損を伴った上皮層の変性を惹起する。この構造破綻が再発性角膜上皮びらんの病態基盤となる。
TGFBI 遺伝子では変異部位と置換アミノ酸の違いが臨床像を決定する。R124C は LCD1 を、R124H は顆粒状角膜ジストロフィ II 型(Avellino 型)を、R124L は Reis-Bücklers 角膜ジストロフィを引き起こす5)。わずか 1 つのアミノ酸の違いが沈着物質(アミロイド vs ヒアリン vs 両者)と沈着部位を決定する分子機構は完全には解明されていないが、変異部位がβig-h3 のどのドメインに属するか、および折り畳み安定性への影響が鍵と考えられている。
LCD IIIA では L527R 等の深層優位変異が太いロープ状の格子状線を生じ、上皮障害を伴わない晩発型となる。沈着物の層別局在は βig-h3 の産生細胞(上皮基底細胞)から実質内への分泌・拡散勾配と、変異蛋白の折り畳み安定性の違いにより説明されうる。R124C は折り畳み中間体からアミロイド原線維形成へ向かう経路を優先し、Bowman 層周辺にアミロイドを蓄積させると考えられている5)。一方、L527R 変異は比較的安定なミスフォールド蛋白を形成し、より深い実質層にゆっくりと沈着する。
従来 LCD1 のアミロイド沈着は前部角膜(Bowman 層〜実質浅層)に限局すると考えられてきた。しかし近年の病理学的検討では、デスメ膜近傍の後部角膜にもアミロイド沈着が存在することが示された3)。後部角膜のアミロイド沈着がデスメ膜の接着に影響を及ぼし、白内障手術時のデスメ膜剥離に寄与する可能性がある3)。前部角膜でアミロイド沈着が上皮接着を障害するのと同様の機序が後部でも作用していると示唆されている3)。
旧LCD2(Meretoja 症候群)の原因分子ゲルソリンは細胞質および細胞外の双方に存在し、アクチン結合を介して細胞運動、細胞分裂、アポトーシスに関与する蛋白である。古典的な D187N 変異ではフィンランド型と呼ばれる、角膜格子状沈着と脳神経障害を主とする表現型を呈する11)。スロベニア家族で報告された新規 p.Glu580Lys 変異は G4-G5 ドメイン境界に位置し、負電荷グルタミン酸から正電荷リジンへの置換により静電的反発を生じ、ドメイン間の連結性と安定性を低下させると考えられている2)。変異型ゲルソリンは血漿中でフーリンおよび MT1-MMP により異常切断を受け、8 kDa および 5 kDa のアミロイド前駆体フラグメントを放出する。これらは角膜基質、皮膚、血管壁、末梢神経、腎糸球体に沈着し、Meretoja 症候群に特徴的な多臓器症状を引き起こす2,11)。角膜沈着は他の全身症状に先行することが多く、眼科医が本症を最初に診断する契機となりうる。
TGFBI 遺伝子の de novo 変異による LCD の発生が報告されている1)。家族歴がない症例でも de novo 変異の可能性を考慮し、遺伝子検査による確認が推奨される1)。L509P 変異は稀であるが、Reis-Bücklers 角膜ジストロフィ様から LCD IIIA 様まで多様な表現型を呈する1)。
GSN 遺伝子では従来の p.Asp187Asn/Tyr 変異に加え、新規の p.Glu580Lys 変異が報告され、角膜格子状ジストロフィ、皮膚弛緩、心臓不整脈、腎障害、視神経障害を伴う全身性アミロイドーシスを呈することが示された2)。
LCD1 患者の後部角膜にアミロイド沈着が存在し、デスメ膜の接着に影響を及ぼす可能性が病理学的に示された3)。白内障手術をはじめとする内眼手術時のデスメ膜剥離リスクへの注意が必要である。
この知見は LCD1 患者の白内障手術適応評価と術式計画に臨床的含意を持つ。
フェムトセカンドレーザー補助層状角膜切除術(femtosecond laser-assisted lamellar keratectomy, FLK)やフェムトセカンドレーザー補助層状角膜移植術(femtosecond laser-assisted lamellar keratoplasty, FALK)など、より精密な手術手技の開発が進められている12)。これらは切除面の平滑性向上と再現性の高い深度制御により、従来の PTK を補完する選択肢として位置づけられつつある。
TGFBI 変異が常染色体優性の機能獲得型変異であることから、変異アレル特異的な siRNA やアンチセンスオリゴヌクレオチド、CRISPR-Cas9 によるアレル特異的ノックアウトが前臨床研究段階で検討されている。角膜は局所投与が可能で免疫特権を有するため、遺伝子治療のターゲット臓器として有利である。ただし現時点で臨床応用されているものはなく、いずれも今後の長期安全性と有効性の検証が必要である。
TGFBIp や変異ゲルソリンの凝集過程を標的とする低分子化合物、分子シャペロン(Hsp70 誘導薬など)、アミロイド原線維結合阻害薬が基礎研究段階で検討されている。全身性のゲルソリン型アミロイドーシスに対しては、血漿中の変異ゲルソリン切断段階を抑制する薬剤が一部の前臨床試験で評価されている2)。将来的にはこうした分子標的治療が、従来の物理的切除(PTK・角膜移植)に代わる根本的治療として期待される。
質量分析を用いた角膜プロテオーム解析により、LCD1 の沈着物中には TGFBIp のみならず複数の異常蛋白が共沈着する可能性が示されている。将来の臨床応用に向け、これらの共沈着蛋白の病態寄与の解明が進められている。