คำจำกัดความ : โรคกระจกตาเสื่อมทางพันธุกรรม ที่มีการสะสมของอะไมลอยด์ในชั้นสโตรมาของกระจกตา เป็นรูปแบบตาข่าย ตามการจำแนก IC3D ฉบับที่ 2 แบ่งเป็น LCD1 และชนิดย่อย (เดิมคือชนิด 3, 3A, 1/3A และ 4) 4) .
สาเหตุ : การถ่ายทอดทางพันธุกรรมแบบออโตโซมัลโดมิแนนต์จากการกลายพันธุ์ของยีน TGFBI การกลายพันธุ์ที่เป็นตัวแทนของ LCD1 คือ R124C (อาร์จินีน 124 เปลี่ยนเป็นซิสเทอีน) 5) .
ชนิดอื่น : โรคอะไมลอยโดซิสทั้งระบบชนิดเจลโซลิน (Meretoja syndrome, เดิมคือ LCD2) ปัจจุบันจัดเป็นโรคอะไมลอยโดซิสในครอบครัว (GSN-AMYL) แยกต่างหาก 10) .
อาการ : LCD1 เริ่มต้นด้วยการสึกกร่อนของกระจกตา ซ้ำในวัยหนุ่มสาว และดำเนินไปสู่ความขุ่นสีขาวบริเวณกลางกระจกตา ในช่วงอายุ 30-40 ปี
การรักษา : การตัดเนื้อกระจกตา ด้วยแสงเลเซอร์เพื่อการรักษา (PTK ) เป็นทางเลือกแรกสำหรับรอยโรคตื้น ส่วนความขุ่นลึกจะรักษาด้วยการปลูกถ่ายกระจกตา ชั้นหน้าส่วนลึก (DALK ) หรือการปลูกถ่ายกระจกตา ทะลุทะลวง 7) .
การพยากรณ์โรค : การกลับเป็นซ้ำใน graft หลังการปลูกถ่ายกระจกตา เป็นสิ่งที่หลีกเลี่ยงไม่ได้ โดยรายงาน 17.8% ใน 5 ปี และ 56% ใน 15 ปีหลังการปลูกถ่ายกระจกตา ทะลุทะลวง 9) .
โรคกระจกตาเสื่อมแบบตาข่าย (lattice corneal dystrophy, LCD) เป็นโรคกระจกตาเสื่อมทางพันธุกรรม ที่มีการสะสมของอะไมลอยด์ในชั้นสโตรมาของกระจกตา ทำให้เกิดความขุ่นเป็นเส้นแบบตาข่าย ถูกบรรยายครั้งแรกในทศวรรษ 1890 และปัจจุบันจำแนกตาม IC3D ฉบับที่ 2 เป็น LCD1 และชนิดย่อย (เดิมคือชนิด 3, 3A, 1/3A และ 4) 4) .
LCD1, โรคกระจกตา เสื่อมแบบเม็ด, โรคกระจกตา เสื่อม Reis-Bücklers และโรคกระจกตา เสื่อม Thiel-Behnke รวมกันเป็นกลุ่มโรคที่เรียกว่า “โรคกระจกตา เสื่อมที่เกี่ยวข้องกับ TGFBI” ยีนก่อโรค TGFBI (transforming growth factor beta-induced gene) อยู่บนแขนยาวของโครโมโซมคู่ที่ 5 (5q31) และถ่ายทอดแบบออโตโซมัลโดมิแนนต์ โปรตีน TGFBI (TGFBIp, kerato-epithelin, βig-h3) ผลิตโดยเยื่อบุกระจกตา และกระจายทั่วทุกชั้นของกระจกตา ในชั้นสโตรมาของกระจกตา โปรตีนนี้มีส่วนร่วมในการสร้างเส้นใยคอลลาเจน แม้แต่การกลายพันธุ์ในยีนเดียวกันก็สามารถทำให้เกิดการสะสมของสารที่แตกต่างกัน (ไฮยาลินหรืออะไมลอยด์) และลักษณะทางคลินิกที่แตกต่างกันมาก ขึ้นอยู่กับตำแหน่งของการกลายพันธุ์และกรดอะมิโนที่ถูกแทนที่ 5) .
การกลายพันธุ์ที่เป็นตัวแทนของ LCD1 คือ R124C ซึ่งอาร์จินีนที่ตำแหน่ง 124 ถูกแทนที่ด้วยซิสเทอีนในยีน TGFBI ในชนิดแปรผัน LCD IIIA มีรายงานการกลายพันธุ์เช่น L527R
โปรตีนผิดปกติที่สะสมในกระจกตา จะย้อมสีแดงด้วย Congo red และแสดงการหักเหสองแนวสีเขียวแอปเปิลภายใต้กล้องจุลทรรศน์โพลาไรซ์ ซึ่งยืนยันว่าเป็นอะไมลอยด์ การค้นพบนี้เป็นตัวบ่งชี้การวินิจฉัยทางเนื้อเยื่อวิทยาแบบคลาสสิกของโรคอะไมลอยโดซิสตั้งแต่ศตวรรษที่ 196)
ชนิดที่เคยเรียกว่า “โรคกระจกตา เสื่อมแบบร่างแหชนิดที่ 2” เป็นอาการทางตาของโรคอะไมลอยโดซิสทั้งระบบชนิดเจลโซลิน (GSN-AMYL, กลุ่มอาการเมเรโตยา) และในการจำแนก IC3D ปัจจุบันจัดเป็น “โรคอะไมลอยโดซิสในครอบครัว” และแยกการรักษาออกจาก LCD แบบคลาสสิก4,10) กลุ่มอาการนี้ซึ่งอธิบายโดย Meretoja ชาวฟินแลนด์ในปี 1969 เป็นโรคทางพันธุกรรมที่มีลักษณะขุ่นมัวของกระจกตา แบบร่างแห ร่วมกับโรคเส้นประสาทสมองเสื่อมที่ลุกลาม ผิวหนังหย่อนยาน และอาการทางระบบ10,11) เนื่องจากความสำคัญของการแยกความแตกต่างในทางคลินิก บทความนี้จึงรวมทั้งสองอย่างไว้ด้วยกัน
ชนิด ยีน การกลายพันธุ์ที่เป็นตัวแทน อายุที่เริ่มมีอาการ ลักษณะสำคัญ LCD1 (ชนิดคลาสสิก) TGFBI (5q31) R124C 10–20 ปี ความขุ่นเป็นเส้นคู่ในบริเวณรูม่านตา , การสึกกร่อนของเยื่อบุผิวซ้ำ LCD IIIA (ชนิดแปรผัน) TGFBI (5q31) L527R และอื่นๆ หลังอายุ 40 ปี เส้นตาข่ายหนาเหมือนเชือกในชั้นสโตรมาลึก ไม่มีการสึกกร่อนของเยื่อบุผิว ชนิด GSN (Meretoja) GSN (9q34) D187N, p.Glu580Lys2) 30-40 ปี เส้นตาข่ายแนวรัศมีบริเวณรอบนอก, โรคอะไมลอยโดซิสทั้งระบบ
ในญี่ปุ่น โรคจอประสาทตา เสื่อมที่เกี่ยวข้องกับ TGFBI ที่พบบ่อยที่สุดคือชนิดเม็ด II (Avellino, R124H) และ LCD1 พบได้น้อยกว่า อย่างไรก็ตาม เนื่องจากทั้งสองชนิดต่างกันเพียงไม่กี่เบสบนยีน TGFBI เดียวกัน การยืนยันด้วยการตรวจทางพันธุกรรมจึงเป็นที่ต้องการในกรณีที่มีภาพทางคลินิกซ้อนทับกัน ยังไม่มีรายงานความชุกที่แน่นอนของ LCD โดยรวมในญี่ปุ่น แต่ถือว่าค่อนข้างหายากในกลุ่มโรคจอประสาทตา เสื่อม
Q
LCD1 และกลุ่มอาการ Meretoja แตกต่างกันอย่างไร?
A
LCD1 คือการสะสมของอะไมลอยด์เฉพาะที่กระจกตา จากการกลายพันธุ์ของยีน TGFBI เริ่มต้นที่บริเวณรูม่านตา ในช่วงอายุ 10-20 ปี และมักมีภาวะเยื่อบุผิวสึกกร่อนซ้ำๆ ในทางกลับกัน กลุ่มอาการ Meretoja (เดิมคือ LCD2, ชนิด GSN) คืออาการทางตาของโรคอะไมลอยโดซิสทั้งระบบจากการกลายพันธุ์ของยีน GSN (เจลโซลิน) เริ่มต้นที่บริเวณรอบนอกของกระจกตา ในช่วงอายุ 30-40 ปี และความใสของส่วนกลางจะคงอยู่นาน กลุ่มอาการ Meretoja มีอาการทั้งระบบ เช่น ผิวหนังหย่อนคล้อย ใบหน้าเหมือนหน้ากาก โรคเส้นประสาทส่วนปลาย และหัวใจเต้นผิดจังหวะ2,10) ใน IC3D ฉบับที่ 2 กลุ่มอาการ Meretoja ถูกจัดประเภทแยกจากโรคจอประสาทตา เสื่อมชนิดร่างแห4)
Liu Z, et al. An R124C mutation in TGFBI caused lattice corneal dystrophy type I with a variable phenotype in three Chinese families. Mol Vis. 2008. Figure 2. PM
CI D: PMC2443752. License: CC BY.
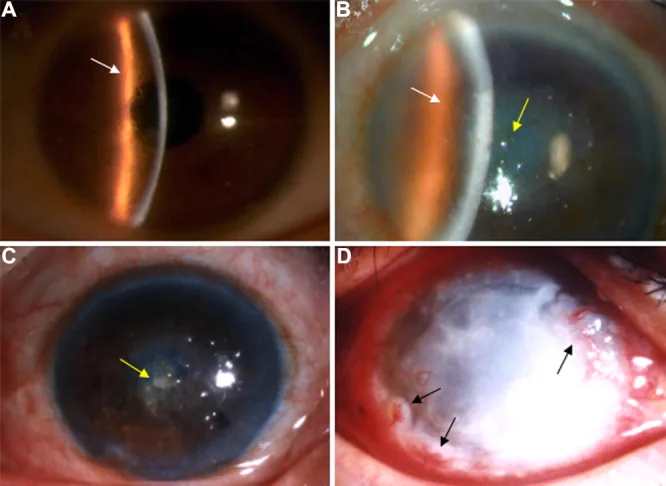
ภาพถ่ายจาก slit lamp แสดงเส้นร่างแหแตกแขนงในชั้นสโตรมาของกระจกตา และความขุ่นที่เด่นชัดบริเวณกลางตา ภาพนี้แสดงลักษณะทางคลินิกที่พบได้ทั่วไปของโรคกระจกตา เสื่อมแบบร่างแห
ใน LCD1 ผู้ป่วยส่วนใหญ่ไม่มีอาการในวัยเด็ก มีเพียงความขุ่นละเอียดที่ตรวจพบได้ด้วยวิธีส่องผ่าน (retroillumination) ใน slit lamp เท่านั้น หลังจากอายุ 10-20 ปี จะเกิดการสึกกร่อนของเยื่อบุกระจกตา ซ้ำๆ (RCE) ร่วมกับอาการปวดตา อย่างรุนแรงเมื่อตื่นนอน กลัวแสง น้ำตาไหล และรู้สึกมีสิ่งแปลกปลอม เมื่ออายุประมาณ 30 ปี จะเกิดความขุ่นสีขาวในชั้นสโตรมาส่วนตื้นบริเวณกลางกระจกตา และการมองเห็น ลดลงหลังจากอายุ 40 ปี
ใน LCD IIIA (ชนิดแปรผัน) มักไม่เกิดความเสียหายต่อเยื่อบุผิว และอาการหลักคือการมองเห็น ลดลงอย่างช้าๆ หลังจากอายุ 40 ปี
ใน LCD2 เดิม (กลุ่มอาการเมเรโตยา) อาการทางตาจะปรากฏในช่วงอายุ 30-40 ปี แต่การสูญเสียการมองเห็น อย่างรุนแรงมักล่าช้าจนถึงอายุ 60 ปี11) อาการทางระบบ เช่น ผิวหนังเปลือกตาหย่อน ใบหน้าเหมือนสวมหน้ากาก โรคเส้นประสาทสมองเสื่อมแบบค่อยเป็นค่อยไป และหัวใจเต้นผิดจังหวะ มักเกิดขึ้นก่อนหรือร่วมด้วย2,10)
ต่อไปนี้คือลักษณะที่พบจากการตรวจด้วย slit lamp ตามชนิดของโรค
LCD1 (ชนิดคลาสสิก)
ตำแหน่งเริ่มแรก : ปรากฏเป็นความขุ่นละเอียดแบบจุดหรือเส้นในบริเวณรูม่านตา ของทั้งสองตา ตั้งแต่ชั้น Bowman ถึงชั้นสโตรมาส่วนตื้น
เส้นร่างแห : ความขุ่นแบบเส้นใยหรือเส้นที่มีขอบสองชั้นพันกันเป็นร่างแหหรือรูปดาว
ระยะลุกลาม : เกิดความขุ่นสีขาวขุ่นรูปไข่หรือกลมบริเวณกระจกตา ส่วนกลาง
การส่องผ่าน (Retroillumination) : เส้นร่างแหบางโปร่งแสงที่มองเห็นได้ยากด้วยการส่องตรงจะปรากฏชัดเจน
การย้อมฟลูออเรสซีน
การสึกกร่อนของเยื่อบุผิวซ้ำ : เกิดขึ้นบ่อยครั้งเนื่องจากสิ่งสะสมขยายไปถึงเซลล์ฐานของเยื่อบุผิวและเยื่อบาวแมน
LCD IIIA (ชนิดแปรผัน)
เส้นร่างแห : เส้นร่างแหหนาและยาวในชั้นสโตรมาชั้นกลางถึงลึก บางครั้งแตกแขนงแบบเดนไดรต์ สามารถสังเกตได้แม้ใช้แสงตรง
ฟีโนไทป์ : มี 3 แบบ: ① เส้นร่างแหอย่างเดียว, ② ตะกอนเม็ดเล็กอย่างเดียว, ③ ผสมทั้งสองแบบ ในบุคคลเดียวกัน ตาขวาและซ้ายอาจมีฟีโนไทป์ต่างกัน และอาจเป็นข้างเดียว
เยื่อบุผิว : ปกติไม่เกิดความเสียหายต่อเยื่อบุผิว
โฮโมไซกัส : ในโฮโมไซกัส L527R เส้นร่างแหจะหนากว่าและตะกอนเม็ดกลางมีขนาดใหญ่กว่า แต่ความแตกต่างไม่เด่นชัดเท่าระหว่างเฮเทอโรไซกัสและโฮโมไซกัส R124H (ชนิดเม็ด II)
ชนิด GSN (Meretoja)
เส้นร่างแห : ตะกอนร่างแหจำนวนเล็กน้อยที่ไม่ละเอียดปรากฏเป็นแนวรัศมีจากส่วนรอบนอก
ความใสส่วนกลาง : ส่วนกลางยังคงใสเป็นเวลานานหลังเริ่มมีอาการ
การกร่อนของเยื่อบุผิว : พบได้น้อย
อาการทั่วร่างกาย : การเปลี่ยนแปลงใบหน้า เช่น หน้าเหมือนหน้ากาก ริมฝีปากยื่นร่วมกับความผิดปกติทางการเคลื่อนไหว หูตก และหนังตาหย่อน 2)
ใน LCD1 ความขุ่นวงกลมกลางอาจรุนแรงในบางราย มีรายงานผู้ป่วยเฮเทอโรไซกัส R124C อายุ 56 ปีที่ได้รับการปลูกถ่ายกระจกตา เนื่องจากความขุ่นวงกลมกลาง
Q
สามารถวินิจฉัย LCD1 ในเด็กได้หรือไม่?
A
LCD1 ในวัยเด็กมักไม่มีอาการ และตรวจพบความผิดปกติได้ยากด้วยแสงตรงเพียงอย่างเดียว การตรวจอย่างละเอียดด้วยกล้องจุลทรรศน์ชีวภาพชนิดกรีดโดยใช้เทคนิคการส่องผ่านหรือการส่องสะท้อนกลับ สามารถสังเกตความขุ่นแบบจุดถึงเส้นเล็กๆ ในชั้นสโตรมาผิวกลาง ในเด็กที่มีการกร่อนเยื่อบุผิวซ้ำ ควรนึกถึง LCD1 โดยซักประวัติครอบครัวและตรวจกระจกตา ของผู้ปกครอง การตรวจยีน TGFBI มีประโยชน์ในการวินิจฉัยที่แน่นอน
ด้านล่างนี้เป็นสรุปยีนก่อโรคและการกลายพันธุ์ทั่วไปของโรคกระจกตา เสื่อมแบบร่างแห
เกี่ยวข้องกับ TGFBI (LCD1, LCD IIIA, LCD IV)
ตำแหน่งยีน : 5q31 (ยีน TGFBI)
รูปแบบการถ่ายทอด : ถ่ายทอดแบบออโตโซมอลโดมิแนนต์
การกลายพันธุ์ที่เป็นตัวแทนของ LCD1 : R124C (Arg124Cys) พบบ่อยที่สุด5)
การกลายพันธุ์ที่เป็นตัวแทนของ LCD IIIA : มีรายงาน L527R (Leu527Arg) และอื่นๆ นอกจากนี้ยังมีกรณีที่เป็นโฮโมไซกัส
การกลายพันธุ์แบบเดอโนโว : มีรายงานกรณีที่มีการกลายพันธุ์แบบเดอโนโว L509P ใน TGFBI ซึ่งแสดงฟีโนไทป์ของ LCD IIIA1) พ่อแม่ไม่มีการกลายพันธุ์ และการกลายพันธุ์ถูกถ่ายทอดไปยังบุตรคนหนึ่ง1)
บทบาทของ TGFBIp : ผลิตโดยเยื่อบุกระจกตา และกระจายทั่วทุกชั้นของกระจกตา มีส่วนร่วมในการสร้างเส้นใยคอลลาเจนในสโตรมา5)
เกี่ยวข้องกับ GSN (กลุ่มอาการเมเรโตยา เดิมคือ LCD2)
ตำแหน่งยีน : 9q34 (ยีน GSN, เจลโซลิน)
รูปแบบการถ่ายทอด : ถ่ายทอดแบบออโตโซมอลโดมิแนนต์
การกลายพันธุ์แบบคลาสสิก : D187N (แบบฟินแลนด์) พบบ่อยที่สุด และยังมีรายงาน p.Asp187Tyr10,11)
การกลายพันธุ์ใหม่ : p.Glu580Lys ที่รายงานในครอบครัวชาวสโลวีเนียอยู่ที่ขอบของโดเมน G4-G5 ทำให้เกิดแรงผลักไฟฟ้าสถิตเนื่องจากการแทนที่ประจุลบด้วยประจุบวก2)
ลักษณะทางคลินิก : นอกจากความขุ่นของกระจกตา แบบตาข่ายแล้ว ยังมีภาวะอะไมลอยโดซิสทั้งระบบร่วมกับผิวหนังหย่อนยาน ภาวะหัวใจเต้นผิดจังหวะ โรคไต และโรคเส้นประสาทตา 2)
เนื่องจากเป็นโรคทางพันธุกรรม ประวัติครอบครัวจึงเป็นปัจจัยเสี่ยงที่สำคัญที่สุด อย่างไรก็ตาม การกลายพันธุ์แบบเดอโนโวใน TGFBI สามารถเกิดขึ้นได้ ดังนั้นการไม่มีประวัติครอบครัวจึงไม่สามารถแยกโรคออกได้1) รูปแบบการถ่ายทอดเป็นแบบออโตโซมอลโดมิแนนต์ หากพ่อหรือแม่คนใดคนหนึ่งเป็นพาหะของการกลายพันธุ์ โอกาสที่จะถ่ายทอดไปยังบุตรคือ 50% ไม่มีความแตกต่างทางเพศ และความแตกต่างทางเชื้อชาติไม่ชัดเจนใน LCD1 แต่กลุ่มอาการเมเรโตยามีชื่อเสียงในเรื่องการสะสมของครอบครัวในฟินแลนด์11)
การมีส่วนร่วมของปัจจัยสิ่งแวดล้อมไม่ชัดเจน และการเริ่มต้นและความก้าวหน้าของโรคโดยพื้นฐานแล้วถูกกำหนดโดยจีโนไทป์ อย่างไรก็ตาม ความถี่ของการสึกกร่อนของเยื่อบุผิวซ้ำอาจแย่ลงในสภาพแวดล้อมที่แห้ง การใช้คอนแทคเลนส์ หรือการบาดเจ็บ การผ่าตัดแก้ไขสายตา (LASIK , SMILE ฯลฯ) อาจทำให้กระจกตา เสื่อมที่เกี่ยวข้องกับ TGFBI แย่ลงอย่างรวดเร็ว และจำเป็นต้องระมัดระวังในกรณีที่มีประวัติครอบครัวระหว่างการคัดกรองก่อนการผ่าตัด5)
เนื่องจากเป็นโรคทางพันธุกรรม จึงไม่มีวิธีการป้องกันโดยตรง สำหรับการป้องกันการสึกกร่อนของเยื่อบุกระจกตา ที่เกิดซ้ำ แนะนำให้ใช้ยาทาขี้ผึ้งตาก่อนนอนหรือน้ำตาเทียม หากมีประวัติครอบครัว ควรไปพบจักษุแพทย์ตั้งแต่เนิ่นๆ และพิจารณาการตรวจทางพันธุกรรม ในเด็กที่มีอาการปวดตา ซ้ำหรือกระจกตา สึกกร่อน การตรวจด้วยกล้องจุลทรรศน์ชนิดกรีด โดยคำนึงถึงโรค dystrophy ที่เกี่ยวข้องกับ TGFBI เป็นสิ่งสำคัญ
เพื่อแยกความแตกต่างระหว่าง LCD1, ชนิดแปรผัน และชนิด GSN จำเป็นต้องรวมผลการตรวจจากกล้องจุลทรรศน์ชนิดกรีด, เนื้อเยื่อวิทยา และพันธุกรรม
การตรวจทางคลินิก
กล้องจุลทรรศน์ชนิดกรีด : ภายใต้แสงส่องตรง เส้นร่างแหในระยะแรกอาจมองข้ามได้ง่าย ด้วยวิธีการส่องผ่าน จะตรวจพบความขุ่นเล็กน้อยบนพื้นหลังของรูม่านตา และด้วยวิธีการส่องย้อนกลับ จะตรวจพบเส้นร่างแหบางโปร่งแสง
การย้อมฟลูออเรสซีน
OCT ส่วนหน้าOCT มีประโยชน์ในการกำหนดความลึกของการตัดออกใน PTK 1)
กล้องจุลทรรศน์คอนโฟคอล ของกระจกตา
การวินิจฉัยที่แน่นอน
การตรวจทางพันธุกรรม : การตรวจหาการกลายพันธุ์ในยีน TGFBI และ GSN ยืนยันชนิดของโรค แม้จะมีฟีโนไทป์เดียวกัน อัตราการกลับเป็นซ้ำและการดำเนินโรคจะแตกต่างกันไปตามการกลายพันธุ์ ซึ่งส่งผลโดยตรงต่อแผนการรักษา
การตรวจทางพยาธิวิทยา : การย้อม Congo Red ให้สีแดง และภายใต้กล้องจุลทรรศน์โพลาไรซ์แสดงการหักเหสองแนวสีเขียวแอปเปิล ยืนยันว่าเป็นอะไมลอยด์6)
อิมมูโนฮิสโตเคมี : สามารถแยกชนิดของโรคได้โดยใช้แอนติบอดีต่อ TGFBIp และแอนติเจลโซลิน
การซักประวัติครอบครัว : เนื่องจากเป็นพันธุกรรมแบบ autosomal dominant การตรวจกระจกตา ของพ่อแม่และพี่น้องจะสนับสนุนการวินิจฉัย
โรค dystrophy กระจกตา แบบเม็ดชนิดที่ 2 (ชนิด Avellino, TGFBI R124H) : พบบ่อยที่สุดในญี่ปุ่นในกลุ่ม dystrophy ที่เกี่ยวข้องกับ TGFBI แสดงการสะสมแบบเม็ดและเส้นร่างแหผสมกัน เพื่อแยกจาก LCD1 การตรวจทางพันธุกรรมมีความแน่นอนโรคอะไมลอยด์กระจกตา ชนิดทุติยภูมิ : ไม่ใช่กรรมพันธุ์ อะไมลอยด์สะสมแบบทุติยภูมิจากการกระตุ้นเรื้อรังของผิวตา เช่น ขนตาคุดหรือกระจกตา รูปกรวย จุดแยกคือไม่มีประวัติครอบครัวและมีโรคพื้นเดิมโรคจอประสาทตา เสื่อมชนิดจุดด่าง (Macular corneal dystrophy) : ถ่ายทอดแบบออโตโซมัลรีเซสซีฟจากการกลายพันธุ์ของยีน CHS T6 ร่วมกับความขุ่นคล้ายกระจกฝ้าฟุ้งกระจายและความผิดปกติของเอ็นโดทีเลียมโรคจอประสาทตา เสื่อมชนิดหยดวุ้น (Gelatinous drop-like corneal dystrophy) : ถ่ายทอดแบบออโตโซมัลรีเซสซีฟจากการกลายพันธุ์ของยีน TACSTD2 มีลักษณะเป็นตุ่มนูนคล้ายวุ้นสีขาวขุ่น พบได้ค่อนข้างบ่อยในญี่ปุ่น
Q
ทำไมการตรวจทางพันธุกรรมจึงสำคัญ?
A
ในโรคจอประสาทตา เสื่อมชนิดร่างแห (lattice corneal dystrophy) แม้ฟีโนไทป์จะคล้ายกัน แต่ยีนก่อโรคและตำแหน่งกลายพันธุ์ที่แตกต่างกันทำให้ความเร็วในการดำเนินโรค ความถี่ของการกลับเป็นซ้ำ ทางเลือกในการรักษา และการมีภาวะแทรกซ้อนทางระบบแตกต่างกันอย่างมาก LCD1 จากการกลายพันธุ์ TGFBI และกลุ่มอาการเมเรโตจา (Meretoja syndrome) จากการกลายพันธุ์ GSN มีความแตกต่างพื้นฐานในแผนการรักษาและความจำเป็นในการตรวจทางระบบ2,10) นอกจากนี้ยังมีรายงานผู้ป่วยที่มีการกลายพันธุ์แบบ de novo ซึ่งประวัติครอบครัวเพียงอย่างเดียวไม่สามารถระบุชนิดของโรคได้1) ดังนั้นการตรวจทางพันธุกรรมจึงจำเป็นสำหรับการวินิจฉัยที่แน่นอนและการจำแนกชนิดของโรค
การรักษาโรคจอประสาทตา เสื่อมชนิดร่างแหขึ้นอยู่กับแนวทางแบบเป็นขั้นตอนตามการจำแนก IC3D
ในวัยเด็กถึงวัยหนุ่มสาว หากไม่มีอาการหรือมีความขุ่นเล็กน้อยเท่านั้น ให้สังเกตอาการ ประเมินการดำเนินโรคด้วยการตรวจด้วยกล้องจุลทรรศน์ชนิดกรีด (slit lamp) ทุก 6 เดือนถึง 1 ปี
สำหรับภาวะกระจกตา ถลอกซ้ำ ซึ่งเป็นอาการหลักของ LCD1 การรักษาแบบประคับประคองต่อไปนี้เป็นขั้นตอนแรก:
การรักษาเมื่อเกิดอาการ : ใช้คอนแทคเลนส์ชนิดนิ่มเพื่อการรักษาแบบสวมต่อเนื่องเพื่อปกป้องเยื่อบุกระจกตา ร่วมกับยาหยอดตาต้านแบคทีเรียเพื่อป้องกันการติดเชื้อทุติยภูมิ ทายาขี้ผึ้งตาเพื่อหล่อลื่นและปกป้องเยื่อบุการป้องกันการกลับเป็นซ้ำ : การทายาขี้ผึ้งตาก่อนนอนมีผลในการยับยั้งการกลับเป็นซ้ำของภาวะกระจกตา ถลอกซ้ำ ในสภาพแวดล้อมที่แห้ง ให้ใช้น้ำตาเทียม หรือสารหล่อลื่นในเวลากลางวันด้วย
ใน LCD1 ที่มีการสะสมของอะไมลอยด์ส่วนใหญ่อยู่ในชั้นผิวของกระจกตา หากมีความขุ่นบริเวณกลางตารุนแรง หรือมีภาวะกระจกตา ถลอกซ้ำ การตัดกระจกตา เพื่อการรักษาด้วยแสงเลเซอร์ (PTK ) โดยใช้เลเซอร์เอกไซเมอร์เป็นทางเลือกแรก7,8) โดยปกติจะไม่เกิดการกลับเป็นซ้ำในระยะแรก แต่การกลับเป็นซ้ำเมื่อเวลาผ่านไปเป็นสิ่งที่หลีกเลี่ยงไม่ได้ และสามารถทำ PTK ในตาเดียวกันได้สูงสุดสองครั้ง
ใน heterozygous การกลับเป็นซ้ำจะช้าและมีเพียงไม่กี่รายที่ต้องรักษาซ้ำ ใน homozygous มักจะกลับเป็นซ้ำเร็วกว่าเมื่อเทียบกับ heterozygous อัตราการกลับเป็นซ้ำหลัง PTK จะเพิ่มขึ้นตามเวลา เช่นเดียวกับ corneal dystrophy ที่เกี่ยวข้องกับ TGFBI อื่นๆ และในการติดตามระยะยาว จะพบสัญญาณของการกลับเป็นซ้ำบางอย่างในผู้ป่วยส่วนใหญ่8) .
ตัวอย่างประสิทธิภาพของ PTK ในผู้ป่วย LCD IIIA จากการกลายพันธุ์ de novo ของ TGFBI L509P ได้ทำ PTK ความลึก 60 µm ภายใต้การนำของ FD-OCT และค่าสายตาที่ดีที่สุดที่แก้ไขแล้ว (BCVA) ดีขึ้นจาก 20/400 เป็น 20/501) ไม่พบการมองเห็น ลดลงหรือการกลับเป็นซ้ำที่มีนัยสำคัญที่ 45 เดือนหลังผ่าตัด1) .
ตามแนวทางปฏิบัติที่พึงประสงค์ของ AAO สำหรับอาการบวมน้ำและความขุ่นของกระจกตา PTK สำหรับ granular และ lattice corneal dystrophy เป็น “ทางเลือกที่สมเหตุสมผล” และอาจชะลอการเปลี่ยนไปใช้ DALK หรือการปลูกถ่ายกระจกตา แบบทะลุทะลวง แต่มีความเสี่ยงต่อการเกิดฝ้าขุ่นหลังผ่าตัด เมื่อทำซ้ำ การใช้ mitomycin C ร่วมกันถือเป็นวิธีการกดการเกิดแผลเป็นซ้ำหรือการสะสมในชั้น stroma และมีการเตือนว่าความเสี่ยงของ corneal ectasia จะเพิ่มขึ้นหากการกร่อนเกินหนึ่งในสามด้านหน้าของ stroma หรือหากเตียงที่เหลือน้อยกว่า 250 µm7) .
ในกรณีที่กลับเป็นซ้ำบ่อยครั้ง หรือความขุ่นลามไปถึงชั้นลึกของ stroma จะเลือกปลูกถ่ายกระจกตา ใน LCD1 โดยทั่วไปจะไม่เหมาะสำหรับการปลูกถ่ายกระจกตา จนกว่าอายุจะเกิน 40 ปี ใน LCD เซลล์เยื่อบุผนังกระจกตา ปกติดังนั้นจึงเลือกเทคนิคตามความลึกของความขุ่น
เทคนิค ข้อบ่งชี้ ลักษณะ การปลูกถ่ายกระจกตา ชั้นผิวความขุ่นตื้น รุกรานน้อย ปฏิเสธน้อย การปลูกถ่ายกระจกตา ชั้นลึก (DALK )ความขุ่นปานกลางถึงลึก รักษาเยื่อบุผนัง ความเสี่ยงปฏิเสธต่ำ การปลูกถ่ายกระจกตา แบบทะลุทะลวง (PK)ความขุ่นตลอดความหนา ฟื้นฟูการมองเห็น สูง แต่เสี่ยงปฏิเสธและกลับเป็นซ้ำ
ในช่วงไม่กี่ปีที่ผ่านมา DALK ถูกใช้อย่างแพร่หลายเป็นทางเลือกแรกใหม่ เนื่องจากลดความเสี่ยงของการปฏิเสธและให้ผลลัพธ์ทางการมองเห็น ที่เทียบเท่ากับการปลูกถ่ายกระจกตา แบบเต็มชั้น
การกลับเป็นซ้ำของ LCD หลังการปลูกถ่ายกระจกตา เป็นสิ่งที่หลีกเลี่ยงไม่ได้ โดยมีรายงานอัตราการกลับเป็นซ้ำหลังการปลูกถ่ายแบบเต็มชั้นที่ 17.8% ใน 5 ปี 26% ใน 8 ปี และ 56% ใน 15 ปี9) เนื่องจากความขุ่นที่กลับเป็นซ้ำมักจำกัดอยู่ที่ชั้นผิว จึงสามารถกำจัดได้ด้วย PTK เพื่อยืดระยะเวลาจนกว่าจะปลูกถ่ายซ้ำ สำหรับ LCD IIIA (ชนิดแปรผัน) มักไม่จำเป็นต้องรักษาเว้นแต่ผลกระทบต่อการมองเห็น จะรุนแรง
ในการผ่าตัดต้อกระจก ในผู้ป่วย LCD1 มีความเสี่ยงต่อการหลุดลอกของเยื่อหุ้มเดสเซเมทเนื่องจากการสะสมของอะไมลอยด์ที่กระจกตา ชั้นหลัง3) ในกรณีหนึ่ง เกิดการหลุดลอกของเยื่อหุ้มเดสเซเมทเป็นบริเวณกว้างระหว่างการทำ I/A ซึ่งจำเป็นต้องปลูกถ่ายกระจกตา แบบเต็มชั้นหลังจาก 8 เดือนเนื่องจากกระจกตา บวมเรื้อรัง3) ในการผ่าตัดภายในลูกตาของผู้ป่วย LCD1 การเฝ้าระวังเยื่อหุ้มเดสเซเมทอย่างระมัดระวังระหว่างการผ่าตัดและการจัดการดูด-ล้างอย่างนุ่มนวลเป็นสิ่งสำคัญ3) สายตายาว หลัง PTK กระจกตา ขุ่น แผลเป็นซ้ำ และกระจกตา โป่งพองเป็นภาวะแทรกซ้อนที่ทราบกันดี7)
Q
PTK มีประสิทธิภาพแค่ไหน?
A
PTK สามารถกำจัดการสะสมของอะไมลอยด์ที่ผิวได้อย่างมีประสิทธิภาพ ช่วยให้การมองเห็น ดีขึ้นและลดการสึกกร่อนของเยื่อบุผิวซ้ำ ในกรณีของ LCD IIIA มีรายงานว่าความคมชัดของการมองเห็น ที่ดีที่สุดแก้ไขแล้วดีขึ้นจาก 20/400 เป็น 20/50 หลัง PTK 60 µm โดยไม่กลับเป็นซ้ำนาน 45 เดือน1) ใน heterozygous การกลับเป็นซ้ำจะช้า แต่ใน homozygous จะเกิดการกลับเป็นซ้ำเร็ว รอยโรคที่ลึกไม่สามารถกำจัดได้ด้วย PTK ดังนั้นความขุ่นลึกจึงจำเป็นต้องทำ DALK หรือปลูกถ่ายกระจกตา แบบเต็มชั้น7)
Q
โรคจะกลับเป็นซ้ำหลังการปลูกถ่ายกระจกตาหรือไม่?
A
การกลับเป็นซ้ำของ LCD หลังการปลูกถ่ายกระจกตา เป็นสิ่งที่หลีกเลี่ยงไม่ได้ อัตราการกลับเป็นซ้ำหลังการปลูกถ่ายแบบเต็มชั้นรายงานที่ 17.8% ใน 5 ปี 26% ใน 8 ปี และ 56% ใน 15 ปี9) อย่างไรก็ตาม ความขุ่นที่กลับเป็นซ้ำมักจำกัดอยู่ที่ชั้นผิวของชิ้นปลูกถ่าย ดังนั้นจึงสามารถกำจัดได้ด้วย PTK เพื่อยืดอายุของชิ้นปลูกถ่าย การปลูกถ่ายกระจกตา แบบชั้นลึก (DALK ) มีความเสี่ยงต่อการปฏิเสธชั้นเอนโดทีเลียมน้อยกว่าการปลูกถ่ายแบบเต็มชั้น และกำลังถูกพิจารณาเป็นทางเลือกแรกใหม่7)
ศูนย์กลางของพยาธิสภาพของ LCD1 คือการสะสมที่ผิดปกติของ TGFBIp (kerato-epithelin, βig-h3) โดยปกติ TGFBIp เป็นโปรตีนโครงสร้างที่ผลิตโดยเยื่อบุผิวกระจกตา และกระจายทั่วทุกชั้นของกระจกตา มีส่วนร่วมในการสร้างเส้นใยคอลลาเจนและการยึดเกาะของเซลล์ในสโตรมา 5) โปรตีนที่ผิดปกติซึ่งเกิดจากการกลายพันธุ์ R124C จะเกิดการพับผิดรูปและการรวมตัวกันเอง ตกตะกอนเป็นเส้นใยอะไมลอยด์ที่ไม่ละลายน้ำในชั้น Bowman ถึงสโตรมาชั้นตื้น ในระยะลุกลาม การสะสมจะแพร่กระจายไปยังสโตรมาชั้นลึก
การสะสมของอะไมลอยด์ทำให้เกิดการเปลี่ยนแปลงในโครงสร้างการยึดเกาะของเยื่อบุผิวกระจกตา ส่วนหน้า ทำให้เกิดการเสื่อมของเซลล์ฐานเยื่อบุผิวและการเสื่อมของชั้นเยื่อบุผิวร่วมกับการขาดหายของเยื่อ Bowman การแตกสลายของโครงสร้างนี้เป็นพื้นฐานทางพยาธิสรีรวิทยาของการสึกกร่อนของเยื่อบุผิวกระจกตา ที่เกิดซ้ำ
ในยีน TGFBI ความแตกต่างของตำแหน่งการกลายพันธุ์และกรดอะมิโนที่ถูกแทนที่จะกำหนดลักษณะทางคลินิก R124C ทำให้เกิด LCD1, R124H ทำให้เกิดกระจกตา เสื่อมชนิดเม็ดแบบที่ II (ชนิด Avellino) และ R124L ทำให้เกิดกระจกตา เสื่อมชนิด Reis-Bücklers 5) กลไกระดับโมเลกุลที่ความแตกต่างของกรดอะมิโนเพียงตัวเดียวกำหนดสารที่สะสม (อะไมลอยด์ vs ไฮยาลิน vs ทั้งสอง) และตำแหน่งที่สะสมยังไม่เป็นที่เข้าใจอย่างสมบูรณ์ แต่เชื่อว่าตำแหน่งของการกลายพันธุ์ในโดเมนใดของ βig-h3 และผลกระทบต่อความเสถียรของการพับตัวเป็นกุญแจสำคัญ
ใน LCD IIIA การกลายพันธุ์ที่เด่นในชั้นลึก เช่น L527R ทำให้เกิดเส้นตาข่ายหนาคล้ายเชือก และเป็นชนิดที่เริ่มมีอาการช้าโดยไม่มีความเสียหายของเยื่อบุผิว การกระจายตัวของตะกอนตามชั้นสามารถอธิบายได้โดยการไล่ระดับการหลั่งและการแพร่ของ βig-h3 จากเซลล์ที่ผลิต (เซลล์ฐานเยื่อบุผิว) ไปยังสโตรมา และความแตกต่างของความเสถียรของการพับตัวของโปรตีนกลายพันธุ์ เชื่อว่า R124C ให้ความสำคัญกับเส้นทางจากตัวกลางการพับตัวไปสู่การสร้างเส้นใยอะไมลอยด์ สะสมอะไมลอยด์รอบชั้น Bowman 5) ในขณะที่การกลายพันธุ์ L527R สร้างโปรตีนที่พับผิดรูปซึ่งค่อนข้างเสถียร และสะสมอย่างช้าๆ ในสโตรมาชั้นลึก
ก่อนหน้านี้เชื่อว่าการสะสมของอะไมลอยด์ใน LCD1 จำกัดอยู่ที่กระจกตา ส่วนหน้า (ชั้น Bowman ถึงสโตรมาชั้นตื้น) อย่างไรก็ตาม การศึกษาทางพยาธิวิทยาเมื่อเร็วๆ นี้แสดงให้เห็นว่ามีการสะสมของอะไมลอยด์ในกระจกตา ส่วนหลังใกล้กับเยื่อหุ้มเดสเซเมทด้วย 3) การสะสมของอะไมลอยด์ในกระจกตา ส่วนหลังอาจส่งผลต่อการยึดเกาะของเยื่อหุ้มเดสเซเมท ซึ่งมีส่วนทำให้เกิดการลอกของเยื่อหุ้มเดสเซเมทระหว่างการผ่าตัดต้อกระจก 3) มีการเสนอว่ากลไกเดียวกับที่ทำให้การยึดเกาะของเยื่อบุผิวในกระจกตา ส่วนหน้าอ่อนแอลงก็ทำงานในส่วนหลังเช่นกัน 3)
โมเลกุลเจลโซลิน (gelsolin) ซึ่งเป็นสาเหตุของกลุ่มอาการเมเรโตยา (Meretoja syndrome) (เดิมคือ LCD2) มีอยู่ทั้งในไซโทพลาซึมและนอกเซลล์ เป็นโปรตีนที่เกี่ยวข้องกับการเคลื่อนที่ของเซลล์ การแบ่งเซลล์ และอะพอพโทซิส ผ่านการจับกับแอคติน การกลายพันธุ์แบบคลาสสิก D187N ซึ่งเรียกว่าแบบฟินแลนด์ ทำให้เกิดการสะสมแบบตาข่ายที่กระจกตา และโรคเส้นประสาทสมองเป็นฟีโนไทป์หลัก11) การกลายพันธุ์ใหม่ p.Glu580Lys ที่รายงานในครอบครัวชาวสโลวีเนียอยู่ที่ขอบเขตโดเมน G4-G5 และการแทนที่กรดกลูตามิกที่มีประจุลบด้วยไลซีนที่มีประจุบวกทำให้เกิดแรงผลักทางไฟฟ้าสถิต ลดการเชื่อมต่อและความเสถียรระหว่างโดเมน2) เจลโซลินที่กลายพันธุ์ถูกตัดอย่างผิดปกติโดยฟูรินและ MT1-MMP ในพลาสมา ปล่อยชิ้นส่วนตั้งต้นของอะไมลอยด์ขนาด 8 kDa และ 5 kDa ชิ้นส่วนเหล่านี้สะสมในสโตรมาของกระจกตา ผิวหนัง ผนังหลอดเลือด เส้นประสาทส่วนปลาย และโกลเมอรูลัสของไต ทำให้เกิดอาการหลายอวัยวะที่เป็นลักษณะเฉพาะของกลุ่มอาการเมเรโตยา2,11) การสะสมที่กระจกตา มักเกิดขึ้นก่อนอาการทางระบบอื่นๆ ดังนั้นจักษุแพทย์อาจเป็นผู้แรกที่วินิจฉัยโรคนี้
มีการรายงานการเกิด LCD เนื่องจากการกลายพันธุ์แบบ de novo ในยีน TGFBI1) แม้ในกรณีที่ไม่มีประวัติครอบครัว ควรพิจารณาความเป็นไปได้ของการกลายพันธุ์แบบ de novo และแนะนำให้ยืนยันด้วยการตรวจทางพันธุกรรม1) การกลายพันธุ์ L509P พบได้น้อย แต่ทำให้เกิดฟีโนไทป์ที่หลากหลายตั้งแต่โรคกระจกตา เสื่อมแบบ Reis-Bücklers ไปจนถึงคล้าย LCD IIIA1)
ในยีน GSN นอกเหนือจากการกลายพันธุ์แบบดั้งเดิม p.Asp187Asn/Tyr แล้ว ยังมีการรายงานการกลายพันธุ์ใหม่ p.Glu580Lys ซึ่งทำให้เกิดอะไมลอยโดซิสทั้งระบบร่วมกับโรคกระจกตาเสื่อมแบบตาข่าย ผิวหนังหย่อนยาน ภาวะหัวใจเต้นผิดจังหวะ โรคไต และโรคเส้นประสาทตา 2)
การศึกษาทางพยาธิวิทยาแสดงให้เห็นว่ามีการสะสมของอะไมลอยด์ในกระจกตา ส่วนหลังของผู้ป่วย LCD1 ซึ่งอาจส่งผลต่อการยึดเกาะของเยื่อหุ้มเดสเซเมท3) จำเป็นต้องระวังความเสี่ยงของการหลุดลอกของเยื่อหุ้มเดสเซเมทระหว่างการผ่าตัดภายในลูกตา เช่น การผ่าตัดต้อกระจก
การค้นพบนี้มีนัยสำคัญทางคลินิกในการประเมินข้อบ่งชี้การผ่าตัดต้อกระจก และการวางแผนเทคนิคการผ่าตัดสำหรับผู้ป่วย LCD1
กำลังมีการพัฒนาวิธีการผ่าตัดที่แม่นยำยิ่งขึ้น เช่น การตัดชั้นกระจกตา โดยใช้เลเซอร์เฟมโตวินาที ช่วย (femtosecond laser-assisted lamellar keratectomy, FLK) และการปลูกถ่ายชั้นกระจกตา โดยใช้เลเซอร์เฟมโตวินาที ช่วย (femtosecond laser-assisted lamellar keratoplasty, FA LK) 12) วิธีการเหล่านี้กำลังถูกวางตำแหน่งให้เป็นทางเลือกเสริมสำหรับ PTK แบบดั้งเดิม เนื่องจากช่วยเพิ่มความเรียบของพื้นผิวที่ตัดและการควบคุมความลึกที่สามารถทำซ้ำได้สูง
เนื่องจากการกลายพันธุ์ TGFBI เป็นการกลายพันธุ์แบบเด่นบนออโตโซมแบบ gain-of-function จึงมีการศึกษา siRNA ที่จำเพาะต่ออัลลีลกลายพันธุ์ โอลิโกนิวคลีโอไทด์ antisense และการน็อกเอาต์แบบจำเพาะอัลลีลด้วย CRISPR-Cas9 ในขั้นตอนการศึกษาก่อนทางคลินิก กระจกตา เป็นอวัยวะเป้าหมายที่ได้เปรียบสำหรับการบำบัดด้วยยีน เนื่องจากสามารถให้ยาเฉพาะที่และมีภูมิคุ้มกันพิเศษ อย่างไรก็ตาม ปัจจุบันยังไม่มีการประยุกต์ใช้ทางคลินิก และทั้งหมดจำเป็นต้องมีการตรวจสอบความปลอดภัยและประสิทธิภาพในระยะยาวในอนาคต
สารประกอบโมเลกุลเล็กที่กำหนดเป้าหมายกระบวนการรวมตัวของ TGFBIp และเจลโซลินกลายพันธุ์ โมเลกุล chaperone (เช่น ตัวเหนี่ยวนำ Hsp70) และยับยั้งการจับกับเส้นใยอะไมลอยด์กำลังถูกศึกษาในขั้นตอนการวิจัยพื้นฐาน สำหรับโรคอะไมลอยโดซิสชนิดเจลโซลินทั่วร่างกาย ยาที่ยับยั้งขั้นตอนการตัดเจลโซลินกลายพันธุ์ในพลาสมาได้รับการประเมินในการศึกษาก่อนทางคลินิกบางส่วน 2) ในอนาคต การบำบัดแบบกำหนดเป้าหมายระดับโมเลกุลเหล่านี้คาดว่าจะมาแทนที่การตัดออกทางกายภาพแบบดั้งเดิม (PTK และการปลูกถ่ายกระจกตา ) ในฐานะการรักษาที่ราก
การวิเคราะห์โปรตีโอมของกระจกตา โดยใช้แมสสเปกโตรเมตรีแสดงให้เห็นว่าสารสะสม LCD1 อาจมีการตกตะกอนร่วมของโปรตีนผิดปกติหลายชนิดนอกเหนือจาก TGFBIp กำลังมีการศึกษาการมีส่วนร่วมทางพยาธิวิทยาของโปรตีนที่ตกตะกอนร่วมเหล่านี้เพื่อการประยุกต์ใช้ทางคลินิกในอนาคต
Ji YW, Ahn H, Shin KJ, Kim TI, Seo KY, Stulting RD, et al. De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. Journal of clinical medicine. 2022;11(11). doi:10.3390/jcm11113055. PMID:35683443; PMCI D:PMC9181583.
Potrc M, Volk M, de Rosa M, Pizem J, Teran N, Jaklic H, Maver A, Drnovsek-Olup B, Bollati M, Vogelnik K, Hocevar A, Gornik A, Pfeifer V, Peterlin B, Hawlina M, Fakin A. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. Int J Mol Sci. 2021;22(3):1084.
Matsumoto A, Fukuoka H, Sotozono C. Descemet’s Membrane Detachment During Cataract Surgery in Lattice Corneal Dystrophy Type I: Histopathological Analysis of Posterior Corneal Involvement. Cureus. 2025;17(3):e81431. doi:10.7759/cureus.81431. PMID:40296953; PMCI D:PMC12037203.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
Lisch W, Seitz B. The clinical landmarks of corneal dystrophies. Developments in ophthalmology. 2011;48:9-23. doi:10.1159/000324075. PMID:21540629.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern. San Francisco, CA: American Academy of Ophthalmology; 2024.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea. 2003;22(1):19-21.
Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969;1(4):314-324. PMID:4313418.
Kivelä T, Tarkkanen A, Frangione B, Ghiso J, Haltia M. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja’s syndrome. Invest Ophthalmol Vis Sci. 1994;35(10):3759-3769. PMID:8088963.
Steger B, Romano V, Biddolph S, Willoughby CE, Batterbury M, Kaye SB. Femtosecond laser-assisted lamellar keratectomy for corneal opacities secondary to anterior corneal dystrophies: an interventional case series. Cornea. 2016;35(1):6-13. PMID:26509759. doi:10.1097/ICO.0000000000000665.