โรคจอตาพร่า ถ่ายทอดทางพันธุกรรมแบบด้อย เกิดจากการกลายพันธุ์ของยีน CHS T6 ที่อยู่บนแขนยาวของโครโมโซมคู่ที่ 16 (16q22)
มีการสะสมของเคราตันซัลเฟตที่ซัลเฟตต่ำ (ไกลโคซามิโนไกลแคน) อย่างกระจายในชั้นสโตรมาของกระจกตา
เป็นหนึ่งในโรคจอตาพร่า หลักสี่ชนิดในญี่ปุ่น (ชนิดเม็ด, ชนิดตาข่าย, ชนิดหยดวุ้น, ชนิดจุด) แต่พบได้ค่อนข้างน้อยในประเทศของเรา
เริ่มมีอาการในช่วงอายุ 10–30 ปี ด้วยการมองเห็น ลดลงทั้งสองข้าง และค่อยๆ เกิดความขุ่นคล้ายกระจกฝ้าที่ลามไปทั่วความหนาของสโตรมา
เมื่อใช้กล้องจุลทรรศน์ชนิดกรีดแสง การสะสมมีลักษณะอยู่ที่ชั้นตื้นบริเวณกลางตาและชั้นลึกบริเวณรอบนอก
ทางภูมิคุ้มกัน แบ่งเป็น 3 ชนิดย่อย: ชนิด I, IA และ II แต่ลักษณะทางคลินิกไม่สามารถแยกความแตกต่างได้
ทางเลือกแรกเมื่อการมองเห็น ลดลงคือการปลูกถ่ายกระจกตา ชั้นหน้าส่วนลึก (DALK ) โดยมีอัตราการกลับเป็นซ้ำประมาณ 5%
เนื่องจากสารผิดปกติยังสะสมที่ชั้นเอ็นโดทีเลียมด้วย การปลูกถ่ายกระจกตา แบบทะลุทะลวง (PKP ) จึงเป็นข้อบ่งชี้ที่ดีสำหรับกรณีที่มีความเสียหายของเอ็นโดทีเลียมขั้นสูง
โรคจอตาพร่า ชนิดจุด (MCD ) เป็นโรคจอตาพร่า ทางพันธุกรรมที่เกิดการสะสมของไกลโคซามิโนไกลแคน (ส่วนใหญ่เป็นเคราตันซัลเฟต) ในชั้นสโตรมาของกระจกตา โรคนี้ถ่ายทอดทางพันธุกรรมแบบด้อย และเกิดจากการกลายพันธุ์ของยีน CHS T6 ที่อยู่บนแขนยาวของโครโมโซมคู่ที่ 16 (16q22)1,3) เดิมเรียกว่าโรคจอตาพร่า Groenouw ชนิด II หรือโรคจอตาพร่า Fehr
แตกต่างจากโรคจอตาพร่า ชั้นสโตรมาอื่นๆ หลายชนิด (ชนิดเม็ด, ชนิดตาข่าย) ที่ถ่ายทอดทางพันธุกรรมแบบเด่น โรคนี้มีลักษณะเด่นคือการถ่ายทอดทางพันธุกรรมแบบ ด้อย ในญี่ปุ่น โรคนี้ถูกนับเป็นหนึ่งในโรคจอตาพร่า หลักสี่ชนิดร่วมกับโรคจอตาพร่า ชนิดเม็ด (ชนิด I และ II), โรคจอตาพร่า ชนิดตาข่าย (ชนิด I และ IIIA) และโรคจอตาพร่า ชนิดหยดวุ้น ซึ่งรวมกันคิดเป็นประมาณ 96% ของโรคจอตาพร่า ทั้งหมด สองชนิดแรกถ่ายทอดทางพันธุกรรมแบบเด่น ส่วนสองชนิดหลัง (ชนิดหยดวุ้นและชนิดจุด) ถ่ายทอดทางพันธุกรรมแบบด้อย
ในการจำแนกประเภท IC3D (คณะกรรมการระหว่างประเทศเพื่อการจำแนกโรคจอตาพร่า เสื่อม) MCD ถูกจัดให้เป็นชนิดหนึ่งของโรคจอตาพร่า เสื่อมชนิดสโตรมา 1) ในบทความปริทัศน์ของ Aggarwal และคณะในวารสาร Survey of Ophthalmology ปี 2018 โรคนี้ถูกอธิบายว่าเป็น “โรคจอตาพร่า เสื่อมชนิดสโตรมาที่พบได้ยากแต่ส่งผลกระทบต่อการมองเห็น อย่างมาก” และได้สรุประบบการวินิจฉัยและการรักษาไว้ 4) ใน IC3D ฉบับที่ 2 โรคจอตาพร่า เสื่อมถูกจำแนกเป็นหมวดหมู่ 1–4 ตามความแข็งแกร่งของหลักฐานทางพันธุกรรม พยาธิวิทยา และลักษณะทางคลินิก 1) MCD ถูกจัดอยู่ใน หมวดหมู่ 1 (โรคจอตาพร่า เสื่อมที่ได้รับการยืนยันในระดับยีน) เนื่องจากการระบุการกลายพันธุ์ของยีน CHS T6
ในทางประวัติศาสตร์ โรคนี้ถูกบรรยายครั้งแรกโดย Groenouw ในปี 1890 ต่อมาเกิดธรรมเนียมการเรียกโรคจอตาพร่า เสื่อมชนิดเม็ดเล็กว่า “ชนิด I” และชนิดจุดด่างว่า “ชนิด II” ในปี 1938 Jones และ Zimmerman ได้กำหนดให้เป็นโรคอิสระ และในปี 2000 Akama และคณะได้อธิบายพื้นฐานระดับโมเลกุลโดยการระบุยีน CHS T6 3)
ทั่วโลกมีความแตกต่างทางภูมิศาสตร์อย่างมาก และในพื้นที่ที่มีความชุกสูงจะพบการรวมตัวในครอบครัว เป็นโรคที่พบได้ค่อนข้างน้อย
ความแตกต่างทางภูมิศาสตร์ของความชุก
สหรัฐอเมริกา : ประมาณ 0.3 ต่อ 250,000 คน พบได้น้อย 2,3)
ไอซ์แลนด์ : ประมาณ 19 ต่อ 250,000 คน เป็นหนึ่งในพื้นที่ที่มีความถี่สูงที่สุดในโลก 5,6)
พื้นที่ที่มีความชุกสูง : อินเดียตอนใต้ ซาอุดีอาระเบีย ไอซ์แลนด์ และสแกนดิเนเวีย 5,7)
พื้นที่อื่นๆ : พบได้ค่อนข้างน้อย เกิดในกรณีการแต่งงานในเครือญาติหรือ heterozygous แบบผสม
ฟีโนไทป์ทางภูมิคุ้มกัน
ชนิด I : ให้ผลลบต่อเคอราตันซัลเฟตในกระจกตา และซีรั่ม 2)
ชนิด IA : ให้ผลบวกภายในเซลล์เคอราโตไซต์ของกระจกตา ให้ผลลบในซีรั่ม 2)
ชนิด II : ให้ผลบวกต่อเคอราตันซัลเฟตในกระจกตา และซีรั่ม 2)
ลักษณะทางคลินิก : ฟีโนไทป์ของทั้งสามชนิดเหมือนกันและไม่สามารถแยกแยะได้ด้วยกล้องสลิตแลมป์ 2,8)
ฟีโนไทป์ทางภูมิคุ้มกันของ MCD ถูกจำแนกตามปริมาณเคอราตันซัลเฟตในกระจกตา และซีรั่มโดยใช้แอนติบอดีโมโนโคลนอลต่อต้านเคอราตันซัลเฟต 2,3)
ฟีโนไทป์ เคราตันซัลเฟตในกระจกตา เคราตันซัลเฟตในซีรัม ชนิด I ลบ ลบ ชนิด IA บวก (ภายในเซลล์) ลบ ชนิด II บวก บวก
ผู้ป่วยส่วนใหญ่จัดอยู่ในชนิด I หรือ IA อย่างไรก็ตาม ทางคลินิก การแยกชนิดย่อยเหล่านี้ไม่สำคัญ และไม่สามารถแยกได้จากการตรวจ2,8) .
Q
โรคจอประสาทตาเสื่อมชนิดจุดด่างแตกต่างจากโรคจอประสาทตาเสื่อมชนิดอื่นอย่างไร?
A
ความแตกต่างที่สำคัญที่สุดคือการถ่ายทอดทางพันธุกรรมแบบออโตโซมอลรีเซสซีฟ ในขณะที่โรคจอประสาทตา เสื่อมชนิดเม็ดและชนิดร่างแหเป็นการถ่ายทอดแบบออโตโซมอลโดมิแนนต์ โรคนี้ต้องการการกลายพันธุ์ในทั้งสองอัลลีลของยีน CHS T6 นอกจากนี้ ยังมีลักษณะขุ่นมัวแบบกระจายคล้ายกระจกฝ้า กระจกตา ทั้งหมดขาวขุ่น และเมื่อตรวจด้วยหลอดกรีด จะพบการสะสมในชั้นตื้นบริเวณกลางและชั้นลึกบริเวณรอบนอก
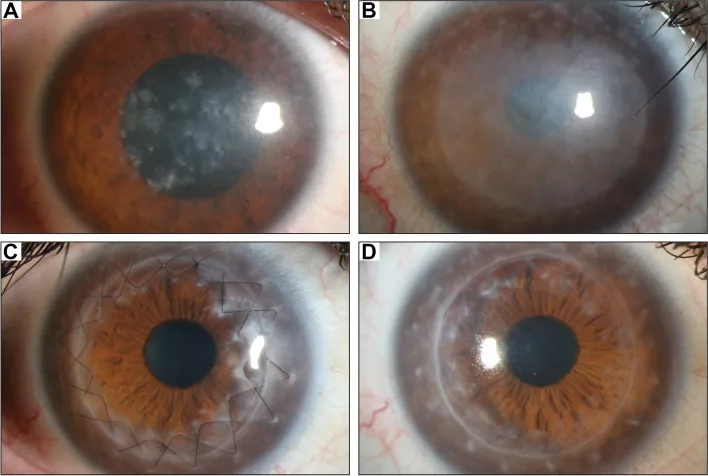
Gassel CJ, et al. Histological findings of corneal tissue after failed phototherapeutic keratectomy in macular corneal dystrophy - a case report. BMC Ophthalmol. 2022. Figure 1. PM
CI D: PMC9080147. License: CC BY.
ภาพถ่ายด้วยกล้อง slit-lamp แสดงความขุ่นกระจายสีเทาขาวและตะกอนเป็นจุดจากส่วนกลางกระจกตา ไปทั่วทั้งกระจกตา แสดงผลการตรวจทางคลินิกโดยทั่วไปของโรค macular corneal dystrophy ช่วยให้เข้าใจความขุ่นของกระจกตา ที่ทำให้การมองเห็น ลดลงได้ง่าย
การมองเห็น ลดลง8) ปวดตา และระคายเคืองกระจกตา ที่เกิดซ้ำกลัวแสง (อาการสู้แสงไม่ได้) : เมื่อความขุ่นของกระจกตา ดำเนินไป การทำงานของการมองเห็น จะลดลงอย่างมากในที่สว่างการสึกกร่อนของเยื่อบุกระจกตา ที่เกิดซ้ำ : อาจเกิดขึ้นซ้ำๆ เนื่องจากความผิดปกติของการยึดเกาะของเยื่อบุกระจกตา
รูปแบบการดำเนินไปโดยทั่วไปของผลการตรวจทางคลินิกมีดังนี้
ทางคลินิก จะพบตะกอนละเอียดกระจายในชั้นสโตรมาของกระจกตา ทำให้เกิดความขุ่นคล้ายกระจกฝ้า เมื่อดำเนินไป ความขุ่นจะขยายไปทั่วทุกชั้นของสโตรมา และกระจายจากส่วนกลางไปยังส่วนรอบนอก หลังจากนั้น นอกจากความขุ่นจางๆ แล้ว จะพบความขุ่นขนาดเล็กจำนวนมากรูปร่างไม่สม่ำเสมอสีเทาขาวในชั้นสโตรมาผิวเผินถึงลึก9)
ผลการตรวจระยะแรก
ความขุ่นแบบจุด : ความขุ่นแบบจุดเล็กสีเทาขาวปรากฏในชั้นสโตรมาผิวเผินของกระจกตา ส่วนกลาง
ความขุ่นคล้ายกระจกฝ้า : พบความขุ่นจางๆ กระจายในชั้นสโตรมาของกระจกตา
ขอบเขตไม่ชัดเจน : ขอบของความขุ่นไม่คมชัด เส้นแบ่งกับสโตรมาปกติไม่ชัดเจน
อาการแสดงระยะลุกลาม
การลุกลามสู่ทุกชั้น : ความขุ่นกระจายไปทั่วทุกชั้นของสโตรมา
การขยายไปสู่ส่วนรอบนอก : ความขุ่นกระจายจากศูนย์กลางไปยังส่วนรอบนอก
กระจกตา บางลงกระจกตา ส่วนกลางลดลง
การสะสมที่เอนโดทีเลียมและเยื่อเดสเซเมท : สารผิดปกติสะสมในโครงสร้างส่วนลึกด้วย
เมื่อตรวจด้วยกล้องจุลทรรศน์ชีวภาพชนิดกรีดแสง (slit-lamp) จะเห็นกระจกตา ทั้งหมดขุ่นแบบกระจาย มีการสะสมเป็นปื้นสีเทาขาวไม่สม่ำเสมอ เมื่อทำภาพตัดขวางด้วยแสงกรีด จะเห็นการกระจายตัวที่มีลักษณะเฉพาะคือ การสะสมอยู่ที่ชั้นตื้นในส่วนกลาง และอยู่ที่ชั้นลึกในส่วนรอบนอก รอยโรคแบบปื้นมักปรากฏเป็นวงกลมซ้อนกันหลายชั้น8) .
ความขุ่นอาจลามไปถึงลิมบัส ซึ่งเป็นจุดสำคัญในการแยกโรคจากกระจกตา เสื่อมชนิดอื่น ในกระจกตา เสื่อมชนิดแกรนูลาร์และชนิดแลตทิซ ลิมบัส มักยังคงใส ในขณะที่ MCD กระจกตา ทั้งหมดมักขุ่นจนถึงลิมบัส 2,8) นอกจากนี้ การเกิดสายตาเอียง ไม่สม่ำเสมอสัมพันธ์กับการสะสมในสโตรมาชั้นหน้า และอาจพบการรับความรู้สึกของกระจกตา ลดลง เนื่องจากการสะสมของสารผิดปกติที่เอนโดทีเลียมเช่นกัน ในรายที่ลุกลามอาจเกิดอาการบวมน้ำของสโตรมาจากการทำงานของเอนโดทีเลียมลดลง8) .
ประวัติธรรมชาติของโรคแตกต่างกันในแต่ละบุคคล แต่มักเป็นไปตามระยะต่อไปนี้
วัยเด็กเล็ก (ระยะไม่มีอาการ) : การกลายพันธุ์ทางพันธุกรรมมีอยู่ตั้งแต่เกิด แต่การตรวจด้วย slit-lamp พบน้อยและไม่มีอาการวัยเรียนถึงวัยรุ่น (ระยะขุ่นเริ่มแรก) : มีความขุ่นกระจายจางๆ ในชั้นตื้นของสโตรมากระจกตา ต่อมาพบการสะสมแบบปื้น10-30 ปี (ระยะการมองเห็น ลดลง) : ความขุ่นดำเนินไป ผู้ป่วยเริ่มสังเกตเห็นการมองเห็น ลดลง30-40 ปี (ระยะลุกลาม) : ความขุ่นกระจายไปทั่วทุกชั้นของสโตรมาและลิมบัส การรับความรู้สึกของกระจกตา ลดลง กระจกตา บางลง และสายตาเอียง ไม่สม่ำเสมอชัดเจนขึ้นวัยกลางคนขึ้นไป (ระยะที่ต้องพิจารณาปลูกถ่ายกระจกตา ) : การทำงานของการมองเห็น ลดลงถึงระดับที่รบกวนชีวิตประจำวัน และพิจารณาการปลูกถ่ายกระจกตา
MCD เป็นโรคที่ดำเนินไปเรื่อยๆ โดยการทำงานของการมองเห็น ลดลงอย่างต่อเนื่องตลอดชีวิต ซึ่งแตกต่างจากกระจกตา เสื่อมชนิดแกรนูลาร์ชนิดที่ 1 และบางชนิดย่อยของกระจกตา เสื่อมชนิดแลตทิซที่ความบกพร่องทางการมองเห็น ไม่รุนแรง4,8) .
ยีนที่เป็นสาเหตุคือ CHS T6 (carbohydrate sulfotransferase 6) 3) อยู่บนโครโมโซม 16q22 และเข้ารหัสเอนไซม์ที่ถ่ายโอนหมู่ซัลเฟตไปยัง N-acetylglucosamine บนโปรตีโอไกลแคนของกระจกตา การกลายพันธุ์ของยีนนี้มีความหลากหลายมาก รวมถึงการกลายพันธุ์แบบ missense, nonsense, frameshift และการขาดหายไปในบริเวณ upstream 5’ ซึ่งมีรายงานในกลุ่มชาติพันธุ์ต่างๆ 3,7)
เนื่องจากการถ่ายทอดทางพันธุกรรมเป็นแบบ autosomal recessive โดยปกติแล้วทั้งพ่อและแม่ของผู้ป่วยเป็นพาหะ อุบัติการณ์มักสูงกว่าในพื้นที่หรือกลุ่มที่มีการแต่งงานในเครือญาติสูง โรคนี้ยังสามารถเกิดขึ้นใน heterozygous compound ที่เกิดจากการแต่งงานระหว่างครอบครัวที่แตกต่างกัน 7)
Akama และคณะในปี 2000 ระบุว่า CHS T6 เป็นยีนก่อโรคนี้ และแสดงให้เห็นว่าทั้งฟีโนไทป์ภูมิคุ้มกันชนิดที่ 1 และชนิดที่ 2 เกิดจากการกลายพันธุ์ที่ตำแหน่งยีนเดียวกัน 3) การค้นพบนี้มีความสำคัญ โดยชี้ให้เห็นว่าความแตกต่างของฟีโนไทป์ภูมิคุ้มกันถูกกำหนดโดยรูปแบบการกลายพันธุ์ที่แตกต่างกันในยีนเดียว และเป็นพื้นฐานสำหรับระบบการวินิจฉัยทางพันธุกรรมในเวลาต่อมา
มีการรายงานการกลายพันธุ์มากกว่า 200 ชนิด โดยการกลายพันธุ์แบบ missense พบบ่อยที่สุด Sultana และคณะระบุการกลายพันธุ์ใหม่จำนวนมากในกลุ่มผู้ป่วยในอินเดียใต้ และแสดงให้เห็นว่าความถี่สูงในภูมิภาคนั้นเกิดจากการสะสมของการกลายพันธุ์เฉพาะพื้นที่ 7) มีรายงานการสะสมในภูมิภาคที่คล้ายกันในซาอุดีอาระเบียและไอซ์แลนด์ และเชื่อว่าภูมิหลังทางประวัติศาสตร์ของประชากร (ผลกระทบจากผู้ก่อตั้งและประเพณีการแต่งงานในเครือญาติ) มีส่วนทำให้เกิดความถี่ของโรค 5,6,7)
ความถี่ของโรคแตกต่างกันไปตามภูมิภาค และเป็นโรคที่ค่อนข้างหายาก 9) เมื่อเทียบกับโรคกระจกตา เสื่อมชนิดอื่น เช่น โรคกระจกตา เสื่อมแบบ granular จำนวนผู้ป่วยน้อยกว่า และมักมีรายงานในครอบครัวที่มีประวัติการแต่งงานในเครือญาติหรือ heterozygous compound
ประวัติครอบครัว : เนื่องจากการถ่ายทอดทางพันธุกรรมเป็นแบบ autosomal recessive ทั้งพ่อและแม่ต้องเป็นพาหะการแต่งงานในเครือญาติ : เพิ่มอัตราการเกิดโรคปัจจัยทางภูมิศาสตร์ : ความชุกสูงในอินเดียใต้ ซาอุดีอาระเบีย ไอซ์แลนด์ และสแกนดิเนเวีย 5,7)
การให้คำปรึกษาทางพันธุกรรม
โรคกระจกตา เสื่อมชนิด macular เป็นโรคทางพันธุกรรมแบบ autosomal recessive หากมีสมาชิกในครอบครัวเป็นโรคเดียวกัน เราขอแนะนำให้รับคำปรึกษาทางพันธุกรรม การวินิจฉัยที่แน่นอนสามารถทำได้โดยการตรวจยีน CHS T6 ซึ่งสามารถดำเนินการได้ในสถานพยาบาลที่ได้รับการรับรอง
Q
จำเป็นต้องตรวจทางพันธุกรรมหรือไม่?
A
การตรวจยีน CHS T6 มีประโยชน์สำหรับการวินิจฉัยที่แน่นอน การตรวจทางพันธุกรรมสามารถดำเนินการได้ในสถานพยาบาลที่ได้รับการรับรอง อย่างไรก็ตาม ในหลายสถานที่ การวินิจฉัยทางคลินิกส่วนใหญ่ขึ้นอยู่กับการตรวจด้วยกล้องจุลทรรศน์ชนิดกรีด (slit-lamp) การตรวจทางพันธุกรรมมีประโยชน์ในการประเมินความเสี่ยงต่อบุตรในอนาคต และเพื่อยืนยันการวินิจฉัยในกรณีที่มีลักษณะทางคลินิกที่ไม่ปกติ
ข้อสงสัย MCD เกิดขึ้นในวัยรุ่นถึงผู้ใหญ่ตอนต้นที่มีความขุ่นกระจายทั่วกระจกตา ทั้งสองข้างและดำเนินไปอย่างต่อเนื่อง ขั้นแรก ซักประวัติโดยละเอียดเกี่ยวกับอาการ (สายตาแย่ลง กลัวแสง ระคายเคือง) ประวัติครอบครัว และการแต่งงานในเครือญาติ จากนั้น ประเมินกระจกตา ด้วยกล้องจุลทรรศน์ชนิดกรีด (slit lamp) ประเมินการทำงานของเซลล์บุผนังกระจกตา และหากจำเป็น ตรวจทางพันธุกรรม
นี่คือการตรวจพื้นฐานสำหรับการวินิจฉัย หากพบความขุ่นของกระจกตา ทั้งสองข้างโดยไม่มีอาการตาแดง หรือบวมน้ำ ให้สงสัยภาวะกระจกตา เสื่อม (corneal dystrophy) ใน MCD จะพบลักษณะดังต่อไปนี้:
ความขุ่นกระจายคล้ายกระจกฝ้า : กระจกตา ทั้งหมดขุ่นอย่างกระจายการสะสมเป็นจุด : จุดขุ่นหลายจุดไม่สม่ำเสมอสีเทาขาวการกระจายแบบศูนย์กลางร่วม : เมื่อตัดด้วยแสงกรีด ชั้นตื้นที่ส่วนกลางและชั้นลึกที่ส่วนปลายการแทรกซึมที่ลิมบัส : ความขุ่นอาจขยายไปถึงลิมบัส ความผิดปกติของผิวเซลล์บุผนังกระจกตา : ในรายที่ลุกลาม อาจพบการสะสมแบบหยดน้ำ
ในขณะที่กระจกตา เสื่อมส่วนใหญ่จะสังเกตเห็นที่ระดับกรีดเป็นรอยโรคที่ไม่ต่อเนื่อง (มีบริเวณใสระหว่างการสะสม) แต่ MCD มีรูปแบบพิเศษคือ รูปแบบความขุ่นกระจาย ร่วมกับกระจกตา เสื่อมแบบร่างแหชนิดที่ 1 และกระจกตา เสื่อมแบบหยดวุ้น ถือเป็นตัวอย่างทั่วไปของ “การสะสมในกระจกตา ที่สังเกตเห็นแบบกระจาย”
เครื่องตรวจวินิจฉัยด้วยแสงความยาวคลื่นใกล้รังสีอินฟราเรดของส่วนหน้าดวงตา (AS-OCT ) : แสดงการกระจายของการสะสมในชั้นตื้นและชั้นลึกของกระจกตา กล้องจุลทรรศน์คอนโฟคอล (in vivo confocal microscopy)8) แผนที่ภูมิประเทศกระจกตา (Corneal topography) : แสดงความหนาแน่นเพิ่มขึ้นที่ยอดกระจกตา และการบางลงของกระจกตา ส่วนกลางกล้องจุลทรรศน์อัลตราซาวนด์ชีวภาพ (UBM ) : มีประโยชน์ในการประเมินความขุ่นลึกและโครงสร้างด้านหลังของกระจกตา กล้องจุลทรรศน์สเปคคูลาร์ : ประเมินความหนาแน่นและสัณฐานวิทยาของเซลล์เอนโดทีเลียม ระดับของรอยโรคที่เอนโดทีเลียมสัมพันธ์โดยตรงกับการเลือกวิธีการผ่าตัดเครื่องเพนทาแคมกระจกตา (เครื่องถ่ายภาพ Scheimpflug) : ให้แผนที่ความหนาแน่นของกระจกตา ทุกชั้น มีประโยชน์ในการประเมินสามมิติของบริเวณที่มีความขุ่น
เพื่อการวินิจฉัยที่แน่นอน การวิเคราะห์ยีน CHS T6 มีประโยชน์ สามารถทำได้ในสถานพยาบาลที่ได้รับการรับรอง
ทางจุลกายวิภาคศาสตร์ การย้อมสี Alcian blue และการย้อมสีเหล็กคอลลอยด์ให้ผลบวก และพบการสะสมแบบกระจายของ glycosaminoglycan ที่มีซัลเฟตต่ำทั้งภายในและภายนอกเซลล์ keratocyte ในชั้นสโตรมาของกระจกตา 2,8) อาจพบการฉีกขาดของเยื่อ Bowman และในกรณีที่ลุกลาม พบสารผิดปกติภายในเซลล์เอนโดทีเลียมด้วย อาจพบลักษณะคล้ายหยดน้ำ (guttae) บนเยื่อ Descemet
โรค รูปแบบการถ่ายทอดทางพันธุกรรม ลักษณะเฉพาะ โรคกระจกตาเสื่อมชนิดเม็ด (Granular corneal dystrophy)autosomal dominant การสะสมแบบเม็ดขอบเขตชัดเจน (มีบริเวณใสคั่นระหว่าง) โรคกระจกตา เสื่อมชนิดร่างแหชนิดที่ 1 (Lattice corneal dystrophy type I) autosomal dominant เส้นร่างแหแบบเส้นตรง (อะไมลอยด์) โรคกระจกตา เสื่อมชนิดเจลาตินัส ดรอปไลค์ ถ่ายทอดแบบออโตโซมอลรีเซสซีฟ รอยโรคยกตัวคล้ายผลหม่อนหรือเป็นแถบ โรคกระจกตา เสื่อมชนิดผลึกของชนีเดอร์ ถ่ายทอดแบบออโตโซมอลโดมิแนนท์ ผลึกรูปเข็ม ร่วมกับภาวะไขมันในเลือดสูง โรคกระจกตา เสื่อมชนิดสโตรมาที่มีมาแต่กำเนิด ถ่ายทอดแบบออโตโซมอลโดมิแนนท์ มีตั้งแต่แรกเกิด สโตรมาหนาตัว โรคเมือกโพลีแซ็กคาไรด์สะสมทั่วร่างกาย (ชนิดกระจกตา ) รีเซสซีฟ/เชื่อมโยงกับโครโมโซม X ร่วมกับอาการทั่วร่างกาย
นอกจากนี้ โรคกระจกตา เสื่อมชนิดโพสทีเรียร์อะมอร์ฟัส (PACD) และโรคกระจกตา เสื่อมชนิดพรีเดสเซเม็ท (PDCD) ก็อยู่ในรายการวินิจฉัยแยกโรคด้วย โรคเมือกโพลีแซ็กคาไรด์สะสมทั่วร่างกาย (เช่น กลุ่มอาการเฮอร์เลอร์, ไช, มอร์ควิโอ) ก็อาจทำให้เกิดความขุ่นของกระจกตา ดังนั้นจึงต้องประเมินรวมถึงอาการทั่วร่างกาย8) .
Q
โรคกระจกตาเสื่อมชนิดแมคคูลาร์วินิจฉัยได้อย่างไร?
A
การวินิจฉัยทางคลินิกทำได้โดยใช้กล้องจุลทรรศน์สลิตแลมป์พบความขุ่นของกระจกตา แบบกระจายคล้ายกระจกฝ้าและตะกอนเป็นจุด ร่วมกับเป็นสองตา ดำเนินโรคเรื้อรัง มีประวัติครอบครัว และอายุที่เริ่มเป็น (10-30 ปี) การตรวจยีน CHS T6 มีประโยชน์ในการยืนยันการวินิจฉัย
เป้าหมายของการรักษา MCD คือ (1) รักษาหรือฟื้นฟูการทำงานของการมองเห็น (2) บรรเทาอาการปวดและอาการระคายเคืองโดยการทำให้ผิวตาคงที่ (3) ป้องกันภาวะแทรกซ้อน (การสึกกร่อนของเยื่อบุผิวและการติดเชื้อ) เนื่องจากไม่มีการรักษาที่สาเหตุของโรค จึงมีการแทรกแซงเป็นขั้นตอนตามระยะและอาการ
ไม่มีการรักษาด้วยยาที่พิสูจน์แล้วว่าชะลอการดำเนินของโรคนี้ 8) มีการดำเนินการดังต่อไปนี้เพื่อบรรเทาอาการ:
น้ำตาเทียม : ปกป้องผิวตาและป้องกันความแห้งสารหล่อลื่น (ยาขี้ผึ้งทาตา) : ปกป้องกระจกตา ในเวลากลางคืนคอนแทคเลนส์ชนิดนอนรักษาโรค : สำหรับจัดการกับการสึกกร่อนของเยื่อบุผิวกระจกตา ที่เกิดซ้ำยาหยอดตาต้านการอักเสบที่ไม่ใช่สเตียรอยด์ (NSAIDs) : ควบคุมความปวด
ในกรณีที่การมองเห็น ลดลงอย่างต่อเนื่อง การปลูกถ่ายกระจกตา เป็นวิธีการรักษาที่หายขาดเพียงวิธีเดียว เทคนิคจะถูกเลือกตามการมีหรือไม่มีโรคของเอ็นโดทีเลียม
การปลูกถ่ายกระจกตา ชั้นหน้าส่วนลึก (DALK )8,9) เนื่องจากการรักษาเอ็นโดทีเลียมของกระจกตา ผู้ป่วยเอง ความเสี่ยงของการปฏิเสธการปลูกถ่ายจึงต่ำกว่าการปลูกถ่ายทะลุทะลวง ในรายงานการประเมินของ AAO (American Academy of Ophthalmology) DALK สำหรับจอตาพร่า ชั้นสโตรมาถูกประเมินว่าให้การฟื้นฟูการมองเห็น ที่เทียบเท่ากับ PKP โดยสูญเสียเอ็นโดทีเลียมน้อยกว่า 8) อัตราการกลับเป็นซ้ำหลัง DALK ต่ำ และการกลับเป็นซ้ำเกิดขึ้นได้ยากเนื่องจากสโตรมาของผู้รับถูกแทนที่การปลูกถ่ายกระจกตา ทะลุทะลวง (PKP )กระจกตา ส่วนกลางบางมาก 7) อายุเฉลี่ยสำหรับ PKP ครั้งแรกสำหรับ MCD คือ 30-40 ปี 7) และอัตราการรอดชีวิตของเนื้อเยื่อปลูกถ่ายดีการตัดกระจกตา ด้วยแสงเพื่อการรักษา (PTK ) : ดำเนินการตามอาการสำหรับการสึกกร่อนของเยื่อบุผิวที่เกิดซ้ำหรือความขุ่นของแผลเป็นชั้นผิว อย่างไรก็ตาม ต้องระวังการเกิดสายตายาว และการเหนี่ยวนำให้เกิดความขุ่นของสโตรมา
ใน MCD สารผิดปกติสามารถสะสมในเซลล์เอ็นโดทีเลียมได้เช่นกัน ดังนั้น ในกรณีที่โรคขยายลึก มักเลือก PKP มากกว่า DALK 7) หากมีความผิดปกติของเอ็นโดทีเลียม การปลูกถ่ายทะลุทะลวงจะเหมาะสม การประเมินเอ็นโดทีเลียมก่อนผ่าตัดอย่างละเอียด (กล้องจุลทรรศน์สเปคคูลาร์หรือคอนโฟคอล) เป็นกุญแจสำคัญในการกำหนดเทคนิคการผ่าตัด
อัตราการกลับเป็นซ้ำหลังการผ่าตัด DALK รายงานว่าต่ำ ประมาณ 5% ในขณะที่อัตราการรอดชีวิตของ graft หลัง PKP ดี มีรายงานหลายฉบับที่แสดงให้เห็นถึงการรอดชีวิตในระยะยาว แต่บางกรณีรายงานว่ามีการสะสมของสารผิดปกติกลับมาบน graft หลังจากหลายปีถึงสิบกว่าปีหลังการผ่าตัด8) ในชุดการศึกษาของ Al-Swailem และคณะในซาอุดีอาระเบีย อัตราการรอดชีวิตของ graft หลัง PKP สำหรับ MCD ดี แต่พบการกลับเป็นซ้ำในบางกรณีระหว่างการติดตามผลระยะยาว8) นอกจากนี้ ในการทบทวนของ AAO โดย Reinhart และคณะ แสดงให้เห็นว่า DALK ให้การฟื้นฟูการมองเห็น ที่เท่าเทียมหรือดีกว่า PKP โดยมีอัตราการสูญเสียเซลล์เยื่อบุผนังหลอดเลือดต่ำ9) การศึกษาหลายศูนย์ของ Unal และคณะยังรายงานประสิทธิภาพของ DALK ในโรค dystrophy ของ stroma รวมถึง MCD
โดยทั่วไป การกลับเป็นซ้ำหลังการปลูกถ่ายกระจกตา เกิดขึ้นเนื่องจากพยาธิวิทยาระดับโมเลกุลของโรคเดิมยังคงอยู่ในฝ่ายผู้รับ ใน DALK เยื่อบุผนังหลอดเลือดและชั้นหน้าของเยื่อหุ้ม Descemet ของผู้รับจะถูกเก็บรักษาไว้ ดังนั้นควรสังเกตว่าโรคอาจดำเนินต่อไปในกรณีที่มีรอยโรคที่เยื่อบุผนังหลอดเลือด หลังการผ่าตัด จำเป็นต้องมีการติดตามผลเป็นระยะในระยะยาว (การตรวจวัดสายตา , การตรวจด้วย slit lamp, การวัดความหนาแน่นของเซลล์เยื่อบุผนังหลอดเลือด)
การปลูกถ่ายกระจกตา
ในโรค dystrophy กระจกตา แบบจุด (macular corneal dystrophy) ความผิดปกติอาจขยายไปถึงเยื่อบุผนังหลอดเลือด ดังนั้นการประเมินเยื่อบุผนังหลอดเลือดก่อนผ่าตัดจึงมีความสำคัญต่อการเลือกเทคนิคการผ่าตัด โรคเดิมอาจกลับเป็นซ้ำหลังการปลูกถ่าย และอาจจำเป็นต้องปลูกถ่ายซ้ำ การติดตามผลหลังการปลูกถ่ายต้องรวมถึงการวัดความหนาแน่นของเซลล์เยื่อบุผนังหลอดเลือดเป็นระยะและการประเมินการกลับเป็นซ้ำของความขุ่น
Q
ควรเลือก DALK หรือ PKP?
A
หากเยื่อบุผนังหลอดเลือดหรือเยื่อหุ้ม Descemet ไม่เกี่ยวข้อง การปลูกถ่ายชั้นหน้าส่วนลึก (DALK ) เป็นทางเลือกแรก DALK เก็บรักษาเยื่อบุผนังหลอดเลือดของกระจกตา ของผู้ป่วยเอง ดังนั้นความเสี่ยงของการปฏิเสธจึงต่ำ และอัตราการกลับเป็นซ้ำรายงานประมาณ 5% หลังการผ่าตัด ในทางกลับกัน ในกรณีที่มีการสะสมของสารผิดปกติในเยื่อบุผนังหลอดเลือดด้วย การปลูกถ่ายกระจกตา แบบเต็มความหนา (PKP ) เป็นข้อบ่งชี้ เทคนิคการผ่าตัดจะพิจารณาจากการประเมินเยื่อบุผนังหลอดเลือดก่อนผ่าตัด
ยีน CHS T6 เข้ารหัส คาร์โบไฮเดรตซัลโฟทรานสเฟอเรส 6 (carbohydrate sulfotransferase 6) 3) เอนไซม์นี้มีหน้าที่ในการถ่ายโอนหมู่ซัลเฟตไปยัง N-acetylglucosamine บนโมเลกุลเคราตาน และจำเป็นสำหรับการสังเคราะห์เคราตานซัลเฟต (KS) ปกติที่อยู่ในโปรตีโอไกลแคนของกระจกตา
เมื่อกิจกรรมของเอนไซม์สูญเสียไปเนื่องจากการกลายพันธุ์ของยีน จะมีการสังเคราะห์เคราตานซัลเฟตที่มีซัลเฟตต่ำและไม่สมบูรณ์ เคราตานซัลเฟตที่ผิดปกตินี้มีความสามารถในการละลายต่ำและสะสมอย่างผิดปกติทั้งภายในและภายนอกเซลล์เคอราโตไซต์ในสโตรมาของกระจกตา 2,3)
ความผิดปกติทั้งในเชิงปริมาณและคุณภาพของเคราตานซัลเฟตนำไปสู่การเปลี่ยนแปลงทางพยาธิวิทยาต่อเนื่องดังต่อไปนี้
การผลิตโปรตีโอไกลแคนขนาดเล็กที่อุดมด้วยลิวซีน (SLRP ) ผิดปกติ : SLRP เฉพาะของกระจกตา เช่น ลูมิแคน (lumican) เคอราโตแคน (keratocan) และไมมีแคน (mimecan) ไม่ถูกสังเคราะห์ตามปกติความผิดปกติของการจัดเรียงเส้นใยคอลลาเจน : SLRP เหล่านี้ควบคุมเส้นผ่านศูนย์กลางของเส้นใยคอลลาเจนของกระจกตา และระยะห่างระหว่างเส้นใยอย่างเคร่งครัด เพื่อรับประกันความโปร่งใส การทำงานของ SLRP ที่ลดลงทำให้เส้นผ่านศูนย์กลางของเส้นใยคอลลาเจนไม่สม่ำเสมอและระยะห่างระหว่างเส้นใยเปลี่ยนแปลงไป 2) การสะสมผิดปกติในเมทริกซ์นอกเซลล์ : เคอราตาน (keratan) ที่ไม่ถูกซัลเฟตจะสะสมตัวในเมทริกซ์นอกเซลล์การกระเจิงแสงเพิ่มขึ้นและการสูญเสียความโปร่งใส : การเปลี่ยนแปลงแบบผสมผสานข้างต้นทำให้การกระเจิงของแสงที่มองเห็นเพิ่มขึ้น ส่งผลให้กระจกตา ทั้งหมดขุ่นมัวแบบกระจาย
การสะสมของไกลโคซามิโนไกลแคน (glycosaminoglycan) สังเกตได้ทั้งภายในและภายนอกเซลล์เคอราโทไซต์ (keratocyte) ของสโตรมา (stroma) และเมื่อโรคดำเนินไป จะแพร่กระจายไปยังเยื่อโบว์แมน (Bowman’s membrane) เยื่อเดสเซเม็ต (Descemet’s membrane) และเซลล์เอนโดทีเลียม (endothelial cells) ในชนิดที่ 1 การลดลงของกิจกรรมเอนไซม์ยังได้รับการยืนยันในกระดูกอ่อนหู (auricular cartilage) ซึ่งบ่งชี้ถึงความเป็นไปได้ว่าเป็นส่วนหนึ่งของความผิดปกติของการเผาผลาญเคอราตานซัลเฟต (keratan sulfate) ทั่วร่างกาย 2) อย่างไรก็ตาม อาการทั่วร่างกายมักไม่ปรากฏทางคลินิก และถือเป็นโรคเฉพาะที่ซึ่งมีอาการหลักที่กระจกตา
ในบรรดาโปรตีโอไกลแคนของกระจกตา ลูมิแคนมีบทบาทในการควบคุมเส้นผ่านศูนย์กลางของเส้นใยคอลลาเจนให้อยู่ที่ประมาณ 25 นาโนเมตร ในขณะที่เคอราโตแคนและไมมีแคนรักษาระยะห่างระหว่างเส้นใยให้สม่ำเสมอ เมื่อสายข้างที่ถูกซัลเฟตของ SLRP เหล่านี้สั้นและไม่สมบูรณ์ เส้นใยคอลลาเจนจะมีความหนาไม่สม่ำเสมอและระยะห่างระหว่างเส้นใยไม่สม่ำเสมอ ผลที่ตามมาคือการกระเจิงแสงภายในสโตรมาของกระจกตา เพิ่มขึ้น และสังเกตเห็นทางคลินิกเป็นความขุ่นคล้ายกระจกฝ้า
ทฤษฎีแลตทิซของเมาเรอร์ (Maurer’s lattice theory) เป็นที่รู้จักในแบบคลาสสิกสำหรับการรักษาความโปร่งใสของกระจกตา ผ่าน “การหักล้างการแทรกสอดของแสงโดยการจัดเรียงแบบแลตทิซที่เป็นระเบียบของเส้นใยคอลลาเจน” แต่ใน MCD โครงสร้างแลตทิซนี้พังทลายลงเนื่องจากความผิดปกติของ SLRP ส่งผลให้สูญเสียความโปร่งใส 2,4)
ในการวิจัยพื้นฐานเมื่อเร็วๆ นี้ มีรายงานว่าความผิดปกติของออโตฟาจี (การกินตัวเอง) ที่เกิดจากการกลายพันธุ์ของ CHS T6 อาจเหนี่ยวนำให้เกิดไพโรพโทซิส (pyroptosis) (การตายของเซลล์แบบอักเสบ) ของเคอราโทไซต์ ซึ่งมีส่วนทำให้โรคดำเนินไป 8) ความผิดปกติของออโตฟาจีที่คล้ายกันนี้ยังมีรายงานในโรคจอประสาทตา เสื่อมชนิดอื่นๆ (เช่น ชนิดเม็ดละเอียด II) ซึ่งกำลังได้รับความสนใจในฐานะกลไกพยาธิวิทยาร่วมของโรคจอประสาทตา เสื่อมโดยทั่วไป
การบำบัดด้วยยีน แบบกำหนดเป้าหมายได้รับการเสนอเป็นกลยุทธ์การรักษาแบบถาวร 8) การวิจัยพื้นฐานเกี่ยวกับการตัดต่อยีนโดยใช้ CRISPR/Cas9 กำลังดำเนินการในโรคจอประสาทตา เสื่อมชนิด Meesmann และอาจกลายเป็นทางเลือกในการรักษาในอนาคตสำหรับ MCD อย่างไรก็ตาม ยังคงมีความท้าทายมากมายสำหรับการประยุกต์ใช้ทางคลินิก เช่น การตัดต่ออัลลีลปกติโดยไม่ได้ตั้งใจ (ผลนอกเป้าหมาย) การพัฒนาวิธีการนำส่งยีนที่มีประสิทธิภาพไปยังเซลล์สโตรมาของกระจกตา และการตรวจสอบความปลอดภัยในระยะยาว
แนวทางการกำจัดเคอราตันซัลเฟตที่ซัลเฟตต่ำซึ่งสะสมในกระจกตา ด้วยเอนไซม์กำลังถูกศึกษาในระดับพื้นฐาน ซึ่งเป็นการประยุกต์แนวคิดการบำบัดด้วยเอนไซม์ทดแทนที่ใช้ในโรคมิวโคโพลีแซ็กคาริโดซิสทั้งระบบมาใช้เฉพาะที่กับกระจกตา แต่ปัจจุบันยังไม่มีรายงานการประยุกต์ใช้ทางคลินิก
การผ่าตัดปลูกถ่ายเยื่อบุผนังกระจกตา แบบ Descemet stripping automated endothelial keratoplasty (DSAEK ) และ Descemet membrane endothelial keratoplasty (DMEK ) เป็นเทคนิคที่พัฒนาขึ้นสำหรับโรคจอประสาทตา เสื่อมชนิด Fuchs และโรคกระจกตาพุพอง เป็นหลัก แต่การประยุกต์ใช้กับกรณี MCD ที่มีรอยโรคที่เยื่อบุผนังกระจกตา ก่อนเป็นหัวข้อวิจัยในอนาคต ปัจจุบัน เนื่องจากโรคนี้ส่งผลกระทบทั้งสโตรมาและเยื่อบุผนังกระจกตา การปลูกถ่ายกระจกตา แบบทะลุทะลวง (PKP ) จึงเป็นทางเลือกที่ปฏิบัติได้ ในอนาคต มีการเสนอแนวคิด “การปลูกถ่ายกระจกตา แบบต่อเนื่อง” ที่รวมการปลูกถ่ายกระจกตา ชั้นหน้าลึก (DALK ) เพื่อแทนที่สโตรมาและการปลูกถ่ายเยื่อบุผนังกระจกตา แบบ DMEK เพื่อแทนที่เยื่อบุผนังกระจกตา เป็นระยะ แต่ทั้งหมดยังอยู่ในขั้นตอนการวิจัย
ระดับเคอราตันซัลเฟตในซีรัมรายงานว่าต่ำกว่าปกติในผู้ป่วย MCD ที่มีฟีโนไทป์ทางภูมิคุ้มกันชนิดที่ 2 และกำลังมีการอภิปรายถึงประโยชน์ของมันในฐานะตัวบ่งชี้เมแทบอลิซึมทั้งระบบ 2) ในอนาคต คาดว่าจะใช้ในการคัดกรองผ่านการตรวจเลือดและการวินิจฉัยตั้งแต่เนิ่นๆ สำหรับพาหะของการกลายพันธุ์ในครอบครัวที่มี CHS T6 นอกจากนี้ การวิเคราะห์ภาพจากกล้องกรีดโดยอัตโนมัติด้วยปัญญาประดิษฐ์ยังสามารถช่วยในการตรวจพบโรคหายากเช่นนี้ได้ตั้งแต่เนิ่นๆ
เนื่องจากโรคนี้เป็นโรคถ่ายทอดทางพันธุกรรมแบบด้อยบนออโตโซม ประมาณ 25% ของพี่น้องของผู้ป่วยอาจได้รับผลกระทบ การใช้การตรวจยีน CHS T6 ทำให้สามารถคัดกรองภายในครอบครัวและให้คำปรึกษาทางพันธุกรรมเกี่ยวกับการแต่งงานและการตั้งครรภ์ได้ แนะนำให้ส่งต่อผู้ป่วยไปยังผู้เชี่ยวชาญตั้งแต่เนิ่นๆ ในครอบครัวที่มีประวัติการแต่งงานในเครือญาติหรือมีกรณีขุ่นมัวของกระจกตา ที่คล้ายกันในครอบครัว
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Klintworth GK. Corneal dystrophies. Orphanet J Rare Dis. 2009;4:7. doi:10.1186/1750-1172-4-7. PMID:19236704; PMCI D:PMC2695576.
Akama TO, Nishida K, Nakayama J, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nature Genetics. 2000;26(2):237-241. doi:10.1038/79987.
Aggarwal S, Peck T, Golen J, Karcioglu ZA. Macular corneal dystrophy: A review. Survey of Ophthalmology. 2018;63(5):609-617. doi:10.1016/j.survophthal.2018.03.004.
Jonasson F, Oshima E, Thonar EJ, et al. Macular corneal dystrophy in Iceland. A clinical, genealogic, and immunohistochemical study of 28 patients. Ophthalmology. 1996;103(7):1111-1117. doi:10.1016/s0161-6420(96)30559-9.
Jonasson F, Johannsson JH, Garner A, Rice NS. Macular corneal dystrophy in Iceland. Eye (London, England). 1989;3 ( Pt 4):446-54. doi:10.1038/eye.1989.66. PMID:2606219.
Sultana A, Sridhar MS , Jagannathan A, et al. Novel mutations of the carbohydrate sulfotransferase-6 (CHS T6) gene causing macular corneal dystrophy in India. Mol Vis. 2003;9:730-734. PMID:14735064.
Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating keratoplasty for macular corneal dystrophy. Ophthalmology. 2005;112(2):220-224. doi:10.1016/j.ophtha.2004.08.017.
Reinhart WJ, Musch DC, Jacobs DS, Lee WB, Kaufman SC, Shtein RM. Deep anterior lamellar keratoplasty as an alternative to penetrating keratoplasty a report by the american academy of ophthalmology. Ophthalmology. 2011;118(1):209-218. doi:10.1016/j.ophtha.2010.11.002. PMID:21199711.