La distrofia maculare corneale (macular corneal dystrophy: MCD) è una distrofia corneale ereditaria caratterizzata dall’accumulo di glicosaminoglicani (principalmente cheratan solfato) nello stroma corneale. Ha una trasmissione autosomica recessiva ed è causata da una mutazione del gene CHST6 situato sul braccio lungo del cromosoma 16 (16q22)1,3). In passato era anche chiamata distrofia corneale di Groenouw tipo II o distrofia corneale di Fehr.
A differenza di molte altre distrofie stromali corneali (granulare, reticolare) che sono autosomiche dominanti, questa malattia è caratterizzata da una trasmissione autosomica recessiva. In Giappone è considerata una delle quattro grandi distrofie corneali insieme alla distrofia granulare (tipo I, II), alla distrofia reticolare (tipo I, IIIA) e alla distrofia gelatinosa a goccia, rappresentando circa il 96% di tutte le distrofie corneali. Di queste, le prime due sono autosomiche dominanti, mentre le ultime due (gelatinosa a goccia e maculare) sono autosomiche recessive.
Secondo la classificazione IC3D (International Committee for Classification of Corneal Dystrophies), la MCD è classificata come un tipo di distrofia stromale 1). In una revisione del 2018 su Survey of Ophthalmology, Aggarwal et al. descrivono questa malattia come «una distrofia stromale rara ma con un impatto significativo sulla funzione visiva» e riassumono il sistema diagnostico e terapeutico 4). Nella seconda edizione della classificazione IC3D, le distrofie corneali sono classificate in categorie da 1 a 4, in base alla forza delle evidenze riguardanti il gene causale, i reperti patologici e il quadro clinico 1). La MCD è classificata come categoria 1 (distrofia stabilita a livello genetico) grazie all’identificazione di mutazioni del gene CHST6.
Storicamente, questa malattia è stata descritta per la prima volta da Groenouw nel 1890, e successivamente è nata la consuetudine di chiamare la distrofia granulare «tipo I» e la distrofia maculare «tipo II». Nel 1938, Jones e Zimmerman la stabilirono come malattia indipendente, e nel 2000 Akama et al. identificarono il gene CHST6, chiarendo la base molecolare 3).
In tutto il mondo esistono notevoli differenze regionali; nelle aree ad alta prevalenza si osserva un accumulo familiare. È una malattia relativamente rara.
Differenze regionali nella prevalenza
Stati Uniti : circa 0,3 persone ogni 250.000 abitanti, rara 2,3)
Islanda : circa 19 persone ogni 250.000 abitanti, una delle regioni più frequenti al mondo 5,6)
Aree ad alta prevalenza : frequenza elevata in India meridionale, Arabia Saudita, Islanda e paesi nordici 5,7)
Altre regioni : relativamente rara. Si verifica in caso di consanguineità o eterozigosi composta
Immunofenotipo
Tipo I : cheratan solfato negativo nella cornea e nel siero 2)
Tipo IA : positivo nei cheratociti corneali, negativo nel siero 2)
Tipo II : cheratan solfato positivo nella cornea e nel siero 2)
Quadro clinico : tutti e tre i tipi hanno lo stesso fenotipo e non sono distinguibili alla lampada a fessura2,8)
L’immunofenotipo della MCD è classificato in base alla quantità di cheratan solfato nella cornea e nel siero utilizzando anticorpi monoclonali anti-cheratan solfato 2,3).
Fenotipo
Cheratan solfato corneale
Cheratan solfato sierico
Tipo I
Negativo
Negativo
Tipo IA
Positivo (intracellulare)
Negativo
Tipo II
Positivo
Positivo
La maggior parte dei pazienti è classificata come tipo I o IA. Tuttavia, clinicamente la distinzione tra questi sottotipi non è importante e non possono essere differenziati all’esame obiettivo2,8).
QIn cosa la distrofia maculare corneale è diversa dalle altre distrofie corneali?
A
La differenza principale è che segue un’ereditarietà autosomica recessiva. Le distrofie granulare e reticolare sono autosomiche dominanti, ma questa malattia richiede mutazioni in entrambi gli alleli del gene CHST6. Inoltre, si presenta con un’opacità diffusa a vetro smerigliato, un offuscamento dell’intera cornea, e alla lampada a fessura i depositi sono superficiali al centro e profondi in periferia.
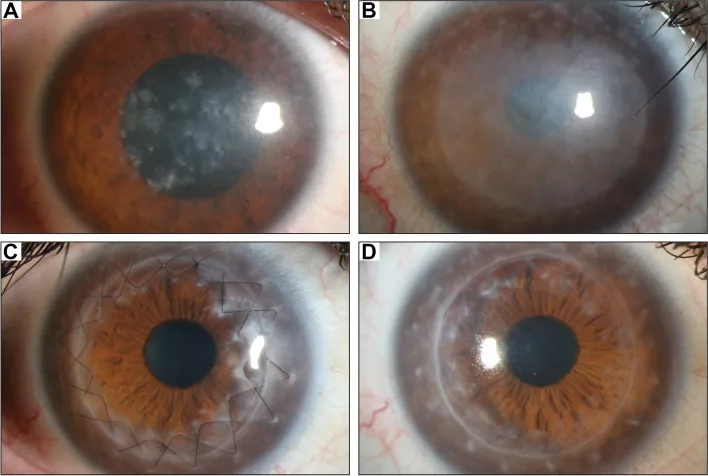
Gassel CJ, et al. Histological findings of corneal tissue after failed phototherapeutic keratectomy in macular corneal dystrophy - a case report. BMC Ophthalmol. 2022. Figure 1. PMCID: PMC9080147. License: CC BY.
Foto con lampada a fessura che mostra un’opacità diffusa grigio-biancastra e depositi maculari dal centro all’intera cornea. Ciò illustra i reperti clinici tipici della distrofia maculare corneale, facilitando la comprensione dell’opacità corneale che causa la riduzione della vista.
Il tipico schema di progressione dei reperti clinici è il seguente.
Clinicamente, si osservano depositi fini diffusi nello stroma corneale, che appare opaco come vetro smerigliato. L’opacità progredisce coinvolgendo tutto lo spessore dello stroma e si estende dal centro alla periferia. Successivamente, oltre all’opacità lieve, compaiono numerose piccole opacità grigio-biancastre di forma irregolare negli strati superficiali e profondi dello stroma.9)
Reperti precoci
Opacità punteggiate : piccole opacità punteggiate grigio-biancastre compaiono negli strati superficiali dello stroma al centro della cornea
Opacità a vetro smerigliato : opacità lieve diffusa dello stroma corneale
Confini sfumati : i bordi dell’opacità sono indistinti, con un confine poco chiaro tra stroma normale e anomalo
Segni di stadio avanzato
Estensione a tutto spessore: l’opacità interessa tutto lo spessore dello stroma
Estensione alla periferia: l’opacità si diffonde dal centro alla periferia
Assottigliamento corneale: lo spessore della cornea centrale diminuisce
Depositi sull’endotelio e sulla membrana di Descemet: sostanze anomale si accumulano anche nelle strutture profonde
Alla lampada a fessura, l’intera cornea è diffusamente opaca con depositi irregolari grigio-biancastri. Al taglio ottico, i depositi sono superficiali al centro e profondi alla periferia, una distribuzione caratteristica. Le lesioni appaiono spesso con pattern concentrici8).
L’opacità può estendersi al limbo, un punto chiave per distinguere la MCD da altre distrofie corneali. Nella distrofia granulare e reticolare, il limbo spesso rimane trasparente, mentre nella MCD l’intera cornea fino al limbo è spesso opaca2,8). L’astigmatismo irregolare è correlato ai depositi stromali anteriori e può verificarsi ipoestesia corneale. I depositi endoteliali possono causare edema stromale per disfunzione endoteliale nei casi avanzati8).
La storia naturale varia da individuo a individuo, ma spesso segue queste fasi.
Prima infanzia (fase asintomatica): la mutazione genetica è presente dalla nascita, ma i reperti alla lampada a fessura sono scarsi e il paziente è asintomatico
Età scolare-adolescenza (fase di opacità precoce): nello stroma anteriore compare una lieve opacità diffusa, poi si osservano depositi maculari
10-30 anni (fase di calo visivo): l’opacità progredisce e il paziente nota un calo della vista
30-40 anni (fase avanzata): l’opacità si estende a tutto lo stroma e al limbo, con ipoestesia corneale, assottigliamento e astigmatismo irregolare evidenti
Età media-avanzata (fase di indicazione al trapianto corneale): la funzione visiva è ridotta al punto da interferire con la vita quotidiana e si considera il trapianto
La MCD è una malattia progressiva con declino continuo della funzione visiva per tutta la vita, distinguendosi dalla distrofia granulare tipo I e reticolare (alcuni sottotipi) dove il deficit visivo rimane lieve4,8).
Il gene responsabile è CHST6 (carbohydrate sulfotransferase 6)3). Situato sul cromosoma 16q22, codifica un enzima che trasferisce un gruppo solfato alla N-acetilglucosamina sui proteoglicani corneali. Le mutazioni di questo gene sono molto numerose: mutazioni missenso, non senso, frameshift, delezioni nella regione 5’ a monte, riportate in vari gruppi etnici3,7).
La trasmissione è autosomica recessiva, quindi entrambi i genitori del probando sono solitamente portatori. L’incidenza è particolarmente elevata in regioni o gruppi con frequenti matrimoni consanguinei. Anche gli eterozigoti composti nati da matrimoni tra famiglie diverse possono sviluppare la malattia7).
Akama et al. nel 2000 hanno identificato CHST6 come gene responsabile, dimostrando che sia l’immunofenotipo I che II sono causati da mutazioni dello stesso locus genico3). Questa scoperta ha suggerito che le differenze nell’immunofenotipo sono determinate da diversi pattern mutazionali di un singolo gene, gettando le basi per la successiva diagnostica genetica.
Sono state riportate oltre 200 mutazioni, le più frequenti sono le missenso. Sultana et al. hanno identificato numerose nuove mutazioni in pazienti dell’India meridionale, dimostrando che l’alta frequenza in quella regione è dovuta a un accumulo di mutazioni specifiche della popolazione7). Accumuli regionali simili sono stati riportati in Arabia Saudita e Islanda, suggerendo che il background storico della popolazione (effetto fondatore, usanze di matrimoni consanguinei) contribuisce alla frequenza della malattia5,6,7).
L’incidenza varia a seconda della regione; è una malattia relativamente rara9). Rispetto ad altre distrofie corneali come la distrofia granulare, i casi sono meno frequenti e spesso riportati in famiglie con eterozigosi composta o background di consanguineità.
Anamnesi familiare: trasmissione autosomica recessiva, entrambi i genitori devono essere portatori
Consanguineità: aumenta l’incidenza
Area geografica: alta prevalenza in India meridionale, Arabia Saudita, Islanda, Scandinavia5,7)
QIl test genetico è necessario?
A
Il test del gene CHST6 è utile per confermare la diagnosi. Può essere eseguito in strutture mediche accreditate. Tuttavia, in molti centri la diagnosi clinica con lampada a fessura rimane predominante. Il test è utile per la valutazione del rischio nei futuri figli o per confermare la diagnosi in casi con presentazione clinica atipica.
Il sospetto di MCD si basa sulla triade: bilateralità, progressione, opacità corneale diffusa in adolescenti o giovani adulti. Innanzitutto si raccoglie in dettaglio l’anamnesi dei sintomi soggettivi (calo visivo, fotofobia, sensazione di irritazione), la storia familiare e la consanguineità. Successivamente, l’esame standard prevede la valutazione della cornea con lampada a fessura, la valutazione della funzione endoteliale e, se necessario, il test genetico.
Questo è l’esame di base per la diagnosi. In presenza di opacità corneale bilaterale senza iperemia o edema corneale, si deve sospettare una distrofia corneale. La MCD presenta i seguenti reperti caratteristici:
Opacità diffusa a vetro smerigliato : l’intera cornea è diffusamente opaca
Depositi maculari : numerose opacità irregolari grigio-biancastre
Distribuzione concentrica : al taglio con la luce a fessura, centralmente superficiali, perifericamente profondi
Infiltrazione limbare : l’opacità può estendersi fino al limbo
Anomalie endoteliali : nei casi avanzati si possono osservare depositi a goccia
Mentre la maggior parte delle distrofie corneali appaiono come lesioni discontinue (con aree trasparenti tra i depositi) all’esame con lampada a fessura, la MCD presenta eccezionalmente un pattern di opacità diffusa. È caratterizzata come uno degli esempi tipici di «depositi corneali osservati diffusamente» insieme alla distrofia corneale reticolare di tipo I e alla distrofia corneale gelatinosa a goccia.
Microscopia confocale in vivo : mostra materiale iperriflettente con margini sfumati e scomparsa dei normali cheratociti8)
Topografia corneale : mostra un aumento della densità all’apice corneale e un assottigliamento corneale centrale
Microscopia ultrasonica biomicroscopica (UBM) : utile per valutare le opacità profonde e la struttura posteriore della cornea
Microscopio speculare : valutazione della densità e morfologia delle cellule endoteliali. Il grado di patologia endoteliale influenza direttamente la scelta della tecnica chirurgica
Pentacam corneale (dispositivo di imaging Scheimpflug) : fornisce una mappa di densità dell’intero spessore corneale, utile per la valutazione tridimensionale delle aree di opacità
Istologicamente, la colorazione con blu Alcian e ferro colloidale è positiva e si osserva un accumulo diffuso di glicosaminoglicani iposolfatati all’interno e all’esterno dei cheratociti dello stroma corneale2,8). Possono verificarsi rotture della membrana di Bowman e, nei casi avanzati, sostanze anomale sono presenti anche nelle cellule endoteliali. Sulla membrana di Descemet possono essere presenti anche aspetti simili a gocce (guttae).
Inoltre, la distrofia corneale posteriore polimorfa (PPCD) e la distrofia corneale pre-Descemet (PDCD) sono incluse nella diagnosi differenziale. Anche le mucopolisaccaridosi sistemiche (sindromi di Hurler, Scheie, Morquio, ecc.) possono causare opacità corneale, pertanto è necessaria una valutazione che includa i reperti sistemici8).
QCome si diagnostica la distrofia maculare della cornea?
A
La diagnosi clinica si basa sull’esame con lampada a fessura che mostra un’opacità corneale diffusa a vetro smerigliato e depositi maculari, la natura bilaterale e progressiva, la storia familiare e l’età di insorgenza (10-30 anni). Il test genetico del gene CHST6 è utile per la conferma.
Gli obiettivi del trattamento della MCD sono: (1) mantenere/recuperare la funzione visiva, (2) alleviare il dolore e i sintomi di irritazione stabilizzando la superficie oculare e (3) prevenire le complicanze (erosione epiteliale, infezione). Poiché non esiste una terapia eziologica, si procede con un intervento graduale in base allo stadio e ai sintomi.
Nei casi di progressivo calo visivo, il trapianto di cornea è l’unico trattamento curativo. La tecnica chirurgica viene scelta in base alla presenza o assenza di coinvolgimento endoteliale.
Cheratoplastica lamellare anteriore profonda (DALK) : trattamento di prima scelta nei casi senza coinvolgimento endoteliale8,9). Preservando l’endotelio corneale del paziente, il rischio di rigetto del trapianto è inferiore rispetto alla cheratoplastica perforante. Anche nel rapporto di valutazione dell’AAO (American Academy of Ophthalmology), la DALK è valutata come equivalente alla PKP nel recupero della funzione visiva con una minore perdita endoteliale per le distrofie stromali8). Il tasso di recidiva dopo DALK è riportato come basso e, poiché lo stroma corneale del ricevente viene sostituito, la recidiva è improbabile.
Cheratoplastica perforante (PKP) : indicata nei casi avanzati con depositi di sostanza anomala nell’endotelio o nella membrana di Descemet, e nei casi di assottigliamento corneale centrale grave7). L’età media per la prima PKP per MCD è riportata tra i 30 e i 40 anni7) e il tasso di sopravvivenza del trapianto è buono.
Cheratectomia fototerapeutica (PTK) : trattamento sintomatico per erosioni epiteliali corneali ricorrenti e opacità cicatriziali superficiali. Tuttavia, è necessario prestare attenzione all’induzione di ipermetropia e opacità stromale.
Poiché nella MCD sostanze anomale possono depositarsi anche nelle cellule endoteliali, quando la malattia si estende in profondità si tende a preferire la PKP rispetto alla DALK7). In caso di anomalia endoteliale, è indicata la cheratoplastica perforante. Una dettagliata valutazione endoteliale preoperatoria (microscopia speculare, confocale) è fondamentale per determinare la tecnica chirurgica.
Il tasso di recidiva dopo DALK è basso, stimato intorno al 5%. La sopravvivenza dell’innesto dopo PKP è buona, con sopravvivenza a lungo termine riportata in molti studi, sebbene siano stati segnalati casi di ri-deposizione di sostanza anomala sull’innesto dopo diversi anni o decenni dall’intervento 8). Nella serie saudita di Al-Swailem et al., la sopravvivenza dopo PKP per MCD era buona, ma in alcuni casi è stata osservata recidiva a lungo termine 8). Inoltre, nella revisione AAO di Reinhart et al., è stato dimostrato che la DALK per le distrofie stromali offre un recupero visivo uguale o superiore alla PKP con un basso tasso di perdita endoteliale 9). Uno studio multicentrico di Unal et al. ha anche riportato l’efficacia della DALK per le distrofie stromali, inclusa la MCD.
In generale, la recidiva postoperatoria degli innesti corneali si verifica perché la patogenesi molecolare della malattia originale persiste nell’ospite. Nella DALK, l’endotelio e lo strato pre-descemetico dell’ospite vengono preservati, quindi si deve notare che la malattia può progredire nei casi con patologia endoteliale. Dopo l’intervento è necessario un follow-up regolare a lungo termine (acuità visiva, lampada a fessura, misurazione della densità delle cellule endoteliali).
QBisogna scegliere DALK o PKP?
A
Se l’endotelio o la membrana di Descemet non sono interessati, la cheratoplastica lamellare anteriore profonda (DALK) è la prima scelta. La DALK preserva l’endotelio corneale del paziente, riducendo il rischio di rigetto, e il tasso di recidiva postoperatoria è di circa il 5%. D’altra parte, nei casi in cui anche l’endotelio presenta depositi anomali, è indicata la cheratoplastica perforante (PKP). La decisione sulla tecnica chirurgica si basa sulla valutazione endoteliale preoperatoria.
6. Fisiopatologia e meccanismo dettagliato della patogenesi
Il gene CHST6 codifica per la carboidrato solfotransferasi 6 3). Questo enzima è responsabile del trasferimento di un gruppo solfato alla N-acetilglucosamina sulla molecola di cheratano ed è essenziale per la normale sintesi del cheratan solfato (KS) presente nei proteoglicani corneali.
La perdita di attività enzimatica dovuta a mutazione genetica porta alla sintesi di un cheratan solfato iposolfatato con solfatazione insufficiente. Questo cheratan solfato anomalo è meno solubile e si deposita in modo anomalo all’interno e all’esterno dei cheratociti dello stroma corneale2,3).
Le anomalie quantitative e qualitative del cheratan solfato portano alla seguente cascata patologica.
Produzione anomala di piccoli proteoglicani ricchi di leucina (SLRP): Gli SLRP specifici della cornea come lumicano, cheratocano e mimecano non vengono sintetizzati normalmente.
Disposizione anomala delle fibre di collagene: Questi SLRP controllano rigorosamente il diametro e la spaziatura delle fibre di collagene corneali, garantendo la trasparenza. La ridotta funzione degli SLRP porta a un diametro eterogeneo delle fibre di collagene e a una modifica della spaziatura2).
Accumulo anomalo nella matrice extracellulare: Il cheratano non solfatato stesso si deposita nella matrice extracellulare.
Aumento della diffusione della luce e perdita di trasparenza: I cambiamenti combinati di cui sopra aumentano la diffusione della luce visibile, e l’intera cornea diventa diffusamente opaca.
L’accumulo di glicosaminoglicani si osserva all’interno e all’esterno dei cheratociti stromali e, con il progredire della lesione, si estende alla membrana di Bowman, alla membrana di Descemet e alle cellule endoteliali. Nel tipo I, una ridotta attività enzimatica è stata confermata anche nella cartilagine dell’orecchio, suggerendo che potrebbe essere una manifestazione parziale di un disturbo sistemico del metabolismo del cheratan solfato2). Tuttavia, clinicamente i sintomi sistemici sono rari e la malattia viene trattata come una condizione localizzata con sintomo principale corneale.
Tra i proteoglicani corneali, il lumicano controlla il diametro delle fibre di collagene a circa 25 nm, mentre il cheratocano e il mimecano mantengono una spaziatura uniforme delle fibre. Quando le catene laterali solfatate di questi SLRP sono corte e incomplete, le fibre di collagene presentano una variazione di diametro e una spaziatura irregolare. Di conseguenza, la diffusione della luce nello stroma corneale aumenta, clinicamente osservata come un’opacità a vetro smerigliato.
Per il mantenimento della trasparenza corneale è classicamente nota la teoria reticolare di Maurer, basata sull’«annullamento dell’interferenza luminosa mediante una disposizione ordinata a reticolo delle fibre di collagene». Nella MCD, questa struttura reticolare viene interrotta dall’anomalia degli SLRP, portando alla perdita di trasparenza2,4).
Recenti ricerche di base hanno riportato che la disfunzione dell’autofagia dovuta alla mutazione CHST6 potrebbe indurre la piroptosi (morte cellulare infiammatoria) dei cheratociti, contribuendo alla progressione della malattia8). Anomalie simili dell’autofagia sono state riportate in altre distrofie corneali (come il tipo granulare II) e stanno attirando l’attenzione come patologia comune a tutte le distrofie corneali.
La terapia genica mirata è proposta come strategia di trattamento permanente 8). La ricerca di base sull’editing genetico utilizzando CRISPR/Cas9 sta progredendo per la distrofia epiteliale corneale di Meesmann e potrebbe diventare un’opzione terapeutica futura anche per la MCD. Tuttavia, rimangono molte sfide per l’applicazione clinica, tra cui modifiche involontarie degli alleli normali (effetti fuori bersaglio), l’istituzione di metodi efficienti di trasferimento genico nelle cellule stromali corneali e la verifica della sicurezza a lungo termine.
Un approccio per rimuovere enzimaticamente il cheratan solfato iposolfatato accumulato nella cornea è studiato a livello fondamentale. Si tratta di applicare localmente alla cornea il concetto di terapia enzimatica sostitutiva già utilizzato nelle mucopolisaccaridosi sistemiche, ma attualmente non ci sono segnalazioni di applicazione clinica.
La cheratoplastica endoteliale con stripping della membrana di Descemet (DSAEK) e la cheratoplastica endoteliale della membrana di Descemet (DMEK) sono tecniche sviluppate principalmente per la distrofia endoteliale di Fuchs e la cheratopatia bollosa, ma la loro applicazione ai casi di MCD con coinvolgimento endoteliale predominante è un argomento di ricerca futura. Attualmente, la cheratoplastica perforante (PKP) è l’opzione realistica per questa malattia che colpisce sia lo stroma che l’endotelio. In futuro, è stato proposto il concetto di «cheratoplastica sequenziale» che combina la DALK (cheratoplastica lamellare anteriore profonda) per sostituire solo lo stroma e la DMEK per sostituire solo l’endotelio, ma queste sono ancora in fase di ricerca.
Il livello sierico di cheratan solfato sarebbe inferiore alla norma nei pazienti con MCD di immunofenotipo II e la sua utilità come marcatore metabolico sistemico è discussa 2). In futuro, si spera in un’applicazione per lo screening tramite esame del sangue e per la diagnosi precoce dei portatori in famiglie con mutazioni CHST6. Inoltre, l’analisi automatizzata delle immagini della lampada a fessura tramite intelligenza artificiale è un’area di ricerca che potrebbe contribuire alla diagnosi precoce di malattie rare come questa.
Poiché si tratta di una malattia autosomica recessiva, circa il 25% dei fratelli di un paziente può essere affetto. L’uso del test genetico CHST6 consente lo screening familiare e la consulenza genetica riguardo al matrimonio e alla gravidanza. Si raccomanda un rinvio precoce a uno specialista nelle famiglie con storia di consanguineità o quando i membri della famiglia presentano opacità corneali simili.
Akama TO, Nishida K, Nakayama J, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nature Genetics. 2000;26(2):237-241. doi:10.1038/79987.
Aggarwal S, Peck T, Golen J, Karcioglu ZA. Macular corneal dystrophy: A review. Survey of Ophthalmology. 2018;63(5):609-617. doi:10.1016/j.survophthal.2018.03.004.
Jonasson F, Oshima E, Thonar EJ, et al. Macular corneal dystrophy in Iceland. A clinical, genealogic, and immunohistochemical study of 28 patients. Ophthalmology. 1996;103(7):1111-1117. doi:10.1016/s0161-6420(96)30559-9.
Jonasson F, Johannsson JH, Garner A, Rice NS. Macular corneal dystrophy in Iceland. Eye (London, England). 1989;3 ( Pt 4):446-54. doi:10.1038/eye.1989.66. PMID:2606219.
Sultana A, Sridhar MS, Jagannathan A, et al. Novel mutations of the carbohydrate sulfotransferase-6 (CHST6) gene causing macular corneal dystrophy in India. Mol Vis. 2003;9:730-734. PMID:14735064.
Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating keratoplasty for macular corneal dystrophy. Ophthalmology. 2005;112(2):220-224. doi:10.1016/j.ophtha.2004.08.017.
Reinhart WJ, Musch DC, Jacobs DS, Lee WB, Kaufman SC, Shtein RM. Deep anterior lamellar keratoplasty as an alternative to penetrating keratoplasty a report by the american academy of ophthalmology. Ophthalmology. 2011;118(1):209-218. doi:10.1016/j.ophtha.2010.11.002. PMID:21199711.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.