حثل القرنية البقعي (MCD) هو حثل قرنية وراثي يتراكم فيه الغليكوز أمينوغليكان (بشكل رئيسي كبريتات الكيراتان) في سدى القرنية. يتبع نمط وراثة جسمي متنحي، وينتج عن طفرة في جين CHST6 الموجود على الذراع الطويل للكروموسوم 16 (16q22)1,3). كان يُسمى سابقًا حثل القرنية من النوع II لجرو نو، أو حثل القرنية لفهر.
على عكس العديد من حثلات سدى القرنية الأخرى (الحبيبي والشبكي) التي تتبع نمط وراثة جسمي سائد، يتميز هذا المرض بكونه وراثيًا جسميًا متنحيًا. في اليابان، يُعد واحدًا من حثلات القرنية الأربعة الكبرى إلى جانب الحثل الحبيبي (النوع I وII)، والحثل الشبكي (النوع I وIIIA)، والحثل الشبيه بالقطيرات الصمغية، وتشكل هذه حوالي 96% من جميع حثلات القرنية. من بينها، الأولان وراثيان جسميان سائدان، والأخيران (الشبيه بالقطيرات الصمغية والبقعي) وراثيان جسميان متنحيان.
في تصنيف IC3D (اللجنة الدولية لتصنيف ضمور القرنية)، يُصنف MCD كنوع من ضمور السدى 1). في مراجعة Aggarwal وآخرين في مجلة Survey of Ophthalmology عام 2018، تم وصف هذا المرض بأنه “ضمور سدى نادر لكنه يؤثر بشكل كبير على الوظيفة البصرية”، وتم تلخيص نظام التشخيص والعلاج 4). في الإصدار الثاني من تصنيف IC3D، تُصنف ضمور القرنية إلى فئات 1-4 بناءً على قوة الأدلة من الجينات المسببة والنتائج المرضية والصورة السريرية 1). يُصنف MCD ضمن الفئة 1 (ضمور مثبت على المستوى الجيني) بسبب تحديد طفرة جين CHST6.
من الناحية التاريخية، تم وصف هذا المرض لأول مرة بواسطة Groenouw في عام 1890، ثم نشأ تقليد تسمية الضمور الحبيبي بـ “النوع الأول” والضمور البقعي بـ “النوع الثاني”. في عام 1938، تم تأسيسه كمرض مستقل بواسطة Jones وZimmerman، وفي عام 2000 تم توضيح الأساس الجزيئي بواسطة Akama وآخرين من خلال تحديد جين CHST6 3).
عالميًا، هناك تباين جغرافي كبير، وفي المناطق ذات الانتشار المرتفع، يُلاحظ تراكم داخل العائلات. إنه مرض نادر نسبيًا.
التباين الجغرافي في الانتشار
الولايات المتحدة: حوالي 0.3 لكل 250,000 شخص، وهو نادر 2,3)
آيسلندا: حوالي 19 لكل 250,000 شخص، وهي واحدة من أعلى المناطق انتشارًا في العالم 5,6)
مناطق الانتشار المرتفع: جنوب الهند، المملكة العربية السعودية، آيسلندا، والدول الاسكندنافية 5,7)
مناطق أخرى: نادرة نسبيًا. يحدث المرض في حالات زواج الأقارب أو التغاير المركب
النمط المناعي
النوع الأول: سلبي لكبريتات الكيراتان في القرنية والمصل 2)
النوع IA: إيجابي داخل الخلايا القرنية (الخلايا القرنية)، سلبي في المصل 2)
النوع الثاني: إيجابي لكبريتات الكيراتان في القرنية والمصل 2)
الصورة السريرية: النمط الظاهري لجميع الأنواع الثلاثة متطابق ولا يمكن تمييزه بواسطة المصباح الشقي2,8)
يُصنف النمط المناعي لـ MCD بناءً على كمية كبريتات الكيراتان في القرنية والمصل باستخدام الأجسام المضادة وحيدة النسيلة المضادة لكبريتات الكيراتان 2,3).
النمط الظاهري
كيراتان سلفات القرنية
كيراتان سلفات المصل
النوع I
سلبي
سلبي
النوع IA
إيجابي (داخل الخلايا)
سلبي
النوع II
إيجابي
إيجابي
معظم المرضى يصنفون ضمن النوع I أو IA. لكن سريريًا، لا أهمية للتمييز بين هذه الأنواع الفرعية، ولا يمكن التفريق بينها بالفحص السريري2,8).
Qما الفرق بين حثل القرنية البقعي وحثلات القرنية الأخرى؟
A
أكبر فرق هو أنه يتبع وراثة جسمية متنحية. بينما حثل القرنية الحبيبي والشبكي يتبعان وراثة جسمية سائدة، يتطلب هذا المرض طفرات في كلا أليلي جين CHST6. كما يتميز بعتامة منتشرة تشبه الزجاج المصنفر، وبياض القرنية بالكامل، وعند الفحص بالمصباح الشقي، توجد ترسبات في الطبقات السطحية في الوسط والطبقات العميقة في المحيط.
Gassel CJ, et al. Histological findings of corneal tissue after failed phototherapeutic keratectomy in macular corneal dystrophy - a case report. BMC Ophthalmol. 2022. Figure 1. PMCID: PMC9080147. License: CC BY.
صورة بالمصباح الشقي تُظهر عتامة منتشرة رمادية بيضاء وترسبات بقعية من مركز القرنية إلى جميع أنحائها. تُظهر النتائج السريرية النموذجية لحثل القرنية البقعي، مما يسهل فهم عتامة القرنية التي تسبب انخفاض الرؤية.
سريريًا، تُرى ترسبات دقيقة منتشرة في سدى القرنية، مما يؤدي إلى عتامة تشبه الزجاج المصنفر. مع تقدم المرض، تمتد العتامة إلى جميع طبقات السدى، وتنتشر من المركز إلى المحيط. بعد ذلك، بالإضافة إلى العتامة الخفيفة، تُرى العديد من العتامات الصغيرة غير المنتظمة ذات اللون الرمادي الأبيض في الطبقات السطحية إلى العميقة من السدى.9)
النتائج المبكرة
عتامة نقطية: تظهر عتامات نقطية صغيرة رمادية بيضاء في الطبقات السطحية من سدى القرنية المركزية
عتامة تشبه الزجاج المصنفر: تُرى عتامة خفيفة منتشرة في سدى القرنية
حدود غير واضحة: حواف العتامة غير حادة، والحدود مع السدى الطبيعي غير واضحة
علامات المرحلة المتقدمة
الامتداد إلى جميع الطبقات: يمتد العتامة إلى جميع طبقات السدى
الامتداد إلى المحيط: ينتشر العتامة من المركز إلى المحيط
ترقق القرنية: يقل سمك القرنية المركزية
ترسبات على البطانة وغشاء دسميه: تتراكم مواد غير طبيعية في البنى العميقة أيضًا
بالمجهر الحيوي، تكون القرنية بأكملها معتمة بشكل منتشر مع ترسبات رمادية-بيضاء غير منتظمة. عند عمل مقطع ضوئي بشق الضوء، يُظهر توزعًا مميزًا حيث تكون الترسبات في الطبقات السطحية في المركز وفي الطبقات العميقة في المحيط. غالبًا ما تظهر الآفات البقعية بنمط دائري متحد المركز8).
قد يمتد العتامة إلى الحوف، وهذه نقطة فارقة مهمة عن ضمورات القرنية الأخرى. في ضمور القرنية الحبيبي والشبكي، غالبًا ما يظل الحوف شفافًا، بينما في MCD، غالبًا ما تكون القرنية بأكملها معتمة حتى الحوف2,8). كما أن ظهور اللابؤرية غير المنتظمة مرتبط بالترسبات في السدى الأمامي، وقد يحدث نقص في حساسية القرنية. نظرًا لترسب المواد غير الطبيعية على البطانة أيضًا، قد يحدث وذمة سدوية بسبب ضعف وظيفة البطانة في الحالات المتقدمة8).
يختلف التطور الطبيعي بين الأفراد، لكنه غالبًا ما يمر بالمراحل التالية.
مرحلة الطفولة المبكرة (مرحلة بدون أعراض): الطفرة الجينية موجودة منذ الولادة، لكن الفحص بالمصباح الشقي يكون محدودًا ولا توجد أعراض
مرحلة المدرسة إلى المراهقة (مرحلة العتامة المبكرة): يظهر عتامة خفيفة منتشرة في الطبقات السطحية من سدى القرنية، ثم تظهر الترسبات البقعية
10-30 سنة (مرحلة انخفاض الرؤية): يتقدم العتامة، ويبدأ المريض في ملاحظة انخفاض الرؤية
30-40 سنة (مرحلة متقدمة): ينتشر العتامة إلى جميع طبقات السدى والحوف، ويصبح نقص حساسية القرنية وترقق القرنية واللابؤرية غير المنتظمة واضحة
منتصف العمر فما فوق (مرحلة الحاجة لزرع القرنية): يصل انخفاض الوظيفة البصرية إلى مستوى يعيق الحياة اليومية، ويتم النظر في زرع القرنية
MCD هو مرض تقدمي يستمر فيه انخفاض الوظيفة البصرية طوال الحياة، وهذا يختلف بشكل كبير عن ضمور القرنية الحبيبي من النوع الأول وبعض الأنواع الفرعية من ضمور القرنية الشبكي حيث يكون ضعف البصر خفيفًا4,8).
الجين المسبب هو CHST6 (carbohydrate sulfotransferase 6)3). يقع على الكروموسوم 16q22 ويشفر إنزيمًا ينقل مجموعة الكبريتات إلى N-أسيتيل جلوكوزامين على بروتيوغليكان القرنية. تتنوع طفرات هذا الجين بشكل كبير، حيث تم الإبلاغ عن طفرات مغلوطة، وطفرات غير منطقية، وطفرات إزاحة الإطار، وحذف في المنطقة المنظمة 5’ في مجموعات عرقية مختلفة 3,7).
نظرًا لأن الوراثة هي صبغية جسدية متنحية، فإن كلا الوالدين عادة ما يكونان حاملين للمرض. يميل معدل الإصابة إلى الارتفاع في المناطق والمجموعات التي يكثر فيها زواج الأقارب. يمكن أن يحدث المرض أيضًا في حالات التغاير المركب الناتجة عن زواج أفراد من عائلات مختلفة 7).
في عام 2000، حدد أكاما وزملاؤه CHST6 كجين مسبب لهذا المرض، وأظهروا أن كلا النمطين المناعيين I و II ينتجان عن طفرات في نفس الموقع الجيني 3). كان هذا الاكتشاف مهمًا يشير إلى أن الاختلاف في النمط المناعي يحدد بواسطة أنماط طفرات مختلفة في جين واحد، وشكل الأساس لنظام التشخيص الجيني اللاحق.
تم الإبلاغ عن أكثر من 200 نوع من الطفرات، وأكثرها شيوعًا هي الطفرات المغلوطة. حددت سلطانة وزملاؤها العديد من الطفرات الجديدة في مجموعة من المرضى في جنوب الهند، وأظهروا أن ارتفاع معدل الإصابة في تلك المنطقة ناتج عن تراكم الطفرات المتركزة محليًا 7). كما تم الإبلاغ عن تراكمات إقليمية مماثلة في المملكة العربية السعودية وأيسلندا، ويُعتقد أن الخلفية التاريخية للسكان (تأثير المؤسس وعادات زواج الأقارب) تساهم في معدل الإصابة 5,6,7).
يختلف معدل الإصابة حسب المنطقة، وهو مرض نادر نسبيًا 9). مقارنة بضمور القرنية الأخرى مثل ضمور القرنية الحبيبي، فإن حالات الإصابة أقل، وتميل إلى الإبلاغ عنها في العائلات التي لديها خلفية من زواج الأقارب أو التغاير المركب.
التاريخ العائلي: نظرًا لأن الوراثة صبغية جسدية متنحية، يجب أن يكون كلا الوالدين حاملين للمرض
زواج الأقارب: يزيد من معدل الإصابة
العامل الجغرافي: ارتفاع معدل الانتشار في جنوب الهند والمملكة العربية السعودية وأيسلندا والدول الاسكندنافية 5,7)
Qهل الاختبار الجيني ضروري؟
A
اختبار جين CHST6 مفيد للتشخيص النهائي. يمكن إجراء الاختبار الجيني في المؤسسات الطبية المعتمدة. ومع ذلك، في معظم المرافق، يعتمد التشخيص السريري بشكل أساسي على فحص المصباح الشقي. الاختبار الجيني مفيد لتقييم خطر إصابة الأطفال في المستقبل، ولتأكيد التشخيص في الحالات غير النمطية.
يبدأ الاشتباه في MCD عندما يكون المريض مراهقًا أو شابًا بالغًا يعاني من عتامة منتشرة في القرنية بأكملها، ثنائية الجانب ومتقدمة. أولاً، يتم أخذ تاريخ مفصل للأعراض الذاتية (انخفاض الرؤية، رهاب الضوء، التهيج)، والتاريخ العائلي، وزواج الأقارب. بعد ذلك، يتم إجراء تقييم للقرنية باستخدام المصباح الشقي، وتقييم وظيفة البطانة، واختبار جيني إذا لزم الأمر.
هذا هو الفحص الأساسي للتشخيص. إذا لوحظ عتامة قرنية ثنائية الجانب دون احمرار أو وذمة، يُشتبه في وجود حثل القرنية. في MCD، تكون النتائج التالية مميزة:
عتامة منتشرة تشبه الزجاج المصنفر: تصبح القرنية بأكملها معتمة بشكل منتشر
ترسبات بقعية: عتامات متعددة غير منتظمة رمادية-بيضاء
توزيع متحد المركز: عند القطع بضوء الشق، تكون الطبقات السطحية في المركز والطبقات العميقة في المحيط
ارتشاح الحوف: قد يمتد العتامة إلى الحوف
شذوذ سطح البطانة: في الحالات المتقدمة، قد تظهر ترسبات قطيرية
بينما تُلاحظ معظم حالات حثل القرنية على مستوى الشق كآفات غير متصلة (مع وجود مناطق شفافة بين الترسبات)، فإن MCD يُظهر بشكل استثنائي نمط عتامة منتشر. إلى جانب حثل القرنية الشبكي من النوع الأول وحثل القرنية القطراتي الجيلاتيني، يُعتبر مثالًا نموذجيًا لـ “ترسبات القرنية التي تُلاحظ بشكل منتشر”.
نسيجيًا، يُظهر صبغة ألسيان بلو وصبغة الحديد الغروي تفاعلًا إيجابيًا، ويُلاحظ تراكم منتشر للجليكوزامينوجليكان منخفض الكبريت داخل وخارج الخلايا القرنية (الخلايا القرنية) 2,8). قد يُلاحظ تمزق في غشاء بومان، وفي الحالات المتقدمة، توجد مواد غير طبيعية داخل الخلايا البطانية. قد تظهر أيضًا زوائد شبيهة بالقطيرات (guttae) على غشاء ديسيميه.
بالإضافة إلى ذلك، يشمل التشخيص التفريقي أيضًا حثل القرنية الخلفي غير المنتظم (PACD) وحثل القرنية أمام غشاء ديسيميه (PDCD). كما أن داء عديد السكاريد المخاطي الجهازي (مثل متلازمات هيرلر وشاي وموركيو) قد يسبب عتامة القرنية، لذا يجب تقييم الأعراض الجهازية8).
Qكيف يتم تشخيص حثل القرنية البقعي؟
A
يتم التشخيص السريري باستخدام المصباح الشقي لملاحظة عتامة القرنية المنتشرة الشبيهة بالزجاج المصنفر والرواسب البقعية، بالإضافة إلى ثنائية العينين والتقدمية والتاريخ العائلي وعمر البداية (10-30 سنة). يُعد اختبار جين CHST6 مفيدًا للتأكيد.
تهدف علاج MCD إلى: (1) الحفاظ على الوظيفة البصرية أو استعادتها، (2) تخفيف الألم وأعراض التهيج عن طريق تثبيت سطح العين، (3) الوقاية من المضاعفات (تآكل الظهارة والعدوى). نظرًا لعدم وجود علاج سببي جذري، يتم التدخل بشكل تدريجي حسب المرحلة والأعراض.
في الحالات المتقدمة مع تدهور الرؤية، يكون زرع القرنية هو العلاج الجذري الوحيد. يتم اختيار التقنية بناءً على وجود مرض بطانة القرنية.
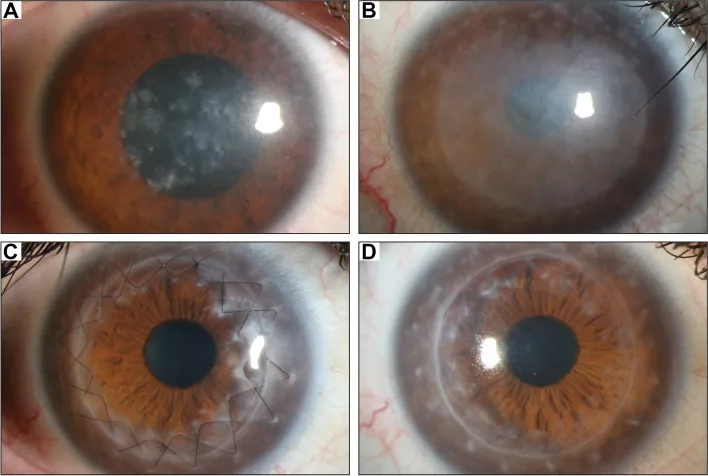
زرع القرنية الأمامي العميق (DALK): هو الخيار الأول في الحالات التي لا تشمل البطانة 8,9). نظرًا للحفاظ على بطانة القرنية الخاصة بالمريض، يكون خطر رفض الطعم أقل من زرع القرنية كامل السمك. في تقرير تقييم الأكاديمية الأمريكية لطب العيون (AAO)، تم تقييم DALK لحثل اللحمة بأنه يحقق استعادة وظيفة بصرية مماثلة لـ PKP مع فقدان أقل للخلايا البطانية 8). معدل التكرار بعد DALK منخفض، والتكرار نادر لأن لحمة القرنية المستقبلة يتم استبدالها.
زرع القرنية كامل السمك (PKP): مناسب للحالات المتقدمة التي تشمل البطانة أو غشاء ديسيميه مع ترسب مواد غير طبيعية، وكذلك الحالات التي يكون فيها مركز القرنية رقيقًا جدًا 7). متوسط عمر أول PKP لـ MCD هو 30-40 سنة 7)، ومعدل بقاء الطعم جيد.
استئصال القرنية العلاجي بالضوء (PTK): يتم إجراؤه بشكل عرضي لتآكل الظهارة المتكرر أو العتامة الندبية السطحية. ومع ذلك، يجب الانتباه إلى تحول القرنية نحو طول النظر وتحريض عتامة اللحمة.
في MCD، يمكن أن تترسب المواد غير الطبيعية أيضًا في الخلايا البطانية، لذلك في الحالات التي يمتد فيها المرض إلى العمق، يميل اختيار PKP على DALK7). في حالة وجود شذوذ في البطانة، يكون زرع القرنية كامل السمك مناسبًا. يعتبر التقييم البطاني التفصيلي قبل الجراحة (بالمجهر المرآوي أو المجهر البؤري) مفتاحًا لتحديد التقنية الجراحية.
تم الإبلاغ عن انخفاض معدل التكرار بعد DALK، حيث يُقدر بحوالي 5%. بينما تكون نسبة بقاء الطعم بعد PKP جيدة، وتحقق العديد من التقارير بقاءً طويل الأمد، إلا أن بعض الحالات أظهرت إعادة ترسب المواد غير الطبيعية على الطعم بعد عدة سنوات إلى عشرات السنين من الجراحة8). في سلسلة Al-Swailem وآخرين في المملكة العربية السعودية، كانت نسبة بقاء الطعم بعد PKP لـ MCD جيدة، ولكن لوحظ تكرار في بعض الحالات خلال المتابعة طويلة الأمد8). كما أظهرت مراجعة Reinhart وآخرين لـ AAO أن DALK يحقق استعادة بصرية مماثلة أو أفضل من PKP مع انخفاض معدل فقدان الخلايا البطانية9). وأظهرت دراسة Unal وآخرين متعددة المراكز فعالية DALK في حالات ضمور السدى بما في ذلك MCD.
بشكل عام، يحدث التكرار بعد زرع القرنية بسبب بقاء الآلية الجزيئية للمرض الأصلي في المضيف. في DALK، يتم الحفاظ على بطانة المضيف والطبقة الأمامية لغشاء ديسيميه، لذلك يجب ملاحظة أن المرض قد يتطور في الحالات التي تعاني من آفات بطانة. بعد الجراحة، تكون المتابعة الدورية طويلة الأمد (فحص حدة البصر، المصباح الشقي، قياس كثافة الخلايا البطانية) ضرورية.
Qأيهما يجب اختياره: DALK أم PKP؟
A
إذا لم تكن البطانة أو غشاء ديسيميه مصابين، فإن زرع الطبقة الأمامية العميقة (DALK) هو الخيار الأول. يحافظ DALK على بطانة القرنية الخاصة بالمريض، مما يقلل من خطر الرفض، ويُبلغ عن معدل تكرار حوالي 5% بعد الجراحة. من ناحية أخرى، في الحالات التي توجد فيها ترسبات مواد غير طبيعية في البطانة أيضًا، يكون زرع القرنية كامل السمك (PKP) مناسبًا. يتم تحديد التقنية الجراحية بناءً على تقييم البطانة قبل الجراحة.
يشفر جين CHST6 إنزيم ناقل سلفات الكربوهيدرات 6 (carbohydrate sulfotransferase 6)3). هذا الإنزيم مسؤول عن نقل مجموعة الكبريتات إلى N-أسيتيل جلوكوزامين على جزيء الكيراتان، وهو ضروري للتخليق الطبيعي لكبريتات الكيراتان (KS) الموجودة في بروتيوغليكان القرنية.
عند فقدان نشاط الإنزيم بسبب طفرة جينية، يتم تخليق كبريتات كيراتان منخفضة الكبريتة وغير مكتملة الكبريتة. هذه الكبريتات الكيراتان غير الطبيعية لها قابلية ذوبان منخفضة وتترسب بشكل غير طبيعي داخل وخارج خلايا الخلايا القرنية (الخلايا القرنية) في سدى القرنية2,3).
تؤدي التشوهات الكمية والنوعية في كبريتات الكيراتان إلى سلسلة من التغيرات المرضية التالية.
إنتاج غير طبيعي للبروتيوغليكانات الصغيرة الغنية بالليوسين (SLRP): عدم تصنيع SLRPs الخاصة بالقرنية مثل اللوميكان والكيراتوكان والميميكان بشكل طبيعي
ترتيب غير طبيعي لألياف الكولاجين: تتحكم هذه SLRPs بشكل صارم في قطر ألياف الكولاجين في القرنية والمسافات بينها، مما يضمن الشفافية. يؤدي انخفاض وظيفة SLRP إلى عدم تجانس قطر ألياف الكولاجين وتغير المسافات بينها 2)
تراكم غير طبيعي في المصفوفة خارج الخلية: يترسب الكيراتان غير المُسلفن نفسه في المصفوفة خارج الخلية
زيادة تشتت الضوء وفقدان الشفافية: تؤدي التغيرات المركبة المذكورة أعلاه إلى زيادة تشتت الضوء المرئي، مما يسبب عتامة منتشرة في القرنية بأكملها
يُلاحظ تراكم الجليكوزامينوجليكان داخل وخلايا الخلايا القرنية (الخلايا اللحمية)، ومع تقدم المرض، ينتشر إلى غشاء بومان وغشاء ديسيميه والخلايا البطانية. في النوع الأول، تم تأكيد انخفاض النشاط الأنزيمي أيضًا في غضروف الأذن، مما يشير إلى احتمال أن يكون جزءًا من اضطراب أيضي جهازي لكبريتات الكيراتان 2). ومع ذلك، نادرًا ما تظهر الأعراض الجهازية سريريًا، ويتم التعامل معه كمرض موضعي تظهر أعراضه الرئيسية في القرنية.
من بين بروتيوغليكانات القرنية، يلعب اللوميكان دورًا في التحكم في قطر ألياف الكولاجين إلى حوالي 25 نانومتر، بينما يحافظ الكيراتوكان والميميكان على تباعد منتظم للألياف. عندما تكون السلاسل الجانبية المُسلفنة لهذه SLRPs قصيرة وغير مكتملة، يحدث تباين في سمك ألياف الكولاجين وتصبح المسافات بينها غير منتظمة. ونتيجة لذلك، يزداد تشتت الضوء داخل سدى القرنية، ويُلاحظ سريريًا على شكل عتامة تشبه الزجاج المصنفر.
تُعرف نظرية ماورر الشبكية (Maurer’s lattice theory) تقليديًا للحفاظ على شفافية القرنية من خلال “إلغاء التداخل الضوئي بواسطة الترتيب الشبكي المنتظم لألياف الكولاجين”، ولكن في MCD، ينهار هذا الهيكل الشبكي بسبب خلل SLRP، مما يؤدي إلى فقدان الشفافية 2,4).
في الأبحاث الأساسية الحديثة، تم الإبلاغ عن أن الخلل الوظيفي في الالتهام الذاتي (البلعمة الذاتية) الناجم عن طفرة CHST6 قد يحفز موت الخلايا المبرمج (الالتهابي) للخلايا القرنية، مما يساهم في تطور المرض 8). تم الإبلاغ عن خلل مماثل في الالتهام الذاتي في حالات ضمور القرنية الأخرى (مثل النوع الحبيبي II)، مما يلفت الانتباه إليه كآلية مرضية شائعة لضمور القرنية بشكل عام.
تم اقتراح العلاج الجيني المستهدف كاستراتيجية علاجية دائمة 8). يجري حاليًا بحث أساسي حول تحرير الجينات باستخدام CRISPR/Cas9 في حثل ظهارة القرنية من نوع ميزمان، وقد يصبح خيارًا علاجيًا مستقبليًا لحثل القرنية المخاطي (MCD). ومع ذلك، لا تزال هناك العديد من التحديات للتطبيق السريري، مثل التحرير غير المقصود للأليلات الطبيعية (تأثيرات خارج الهدف)، وتطوير طرق فعالة لتوصيل الجينات إلى الخلايا اللحمية للقرنية، والتحقق من السلامة طويلة المدى.
يتم دراسة نهج إزالة الكيراتان سلفات منخفض الكبريتة المتراكم في القرنية إنزيميًا على المستوى الأساسي. هذا يطبق مفهوم العلاج بالإنزيمات البديلة المستخدم في داء عديد السكاريد المخاطي الجهازي موضعيًا على القرنية، ولكن لا توجد تقارير عن تطبيقه السريري حتى الآن.
تعد جراحة رأب القرنية البطانية باستخدام نزع غشاء ديسيميه (DSAEK) ورأب القرنية البطانية بغشاء ديسيميه (DMEK) تقنيات تم تطويرها بشكل أساسي لعلاج حثل فوكس البطاني واعتلال القرنية الفقاعي، ولكن تطبيقها على حالات حثل القرنية المخاطي (MCD) التي تصيب البطانة أولاً هو موضوع بحث مستقبلي. حاليًا، نظرًا لأن المرض يصيب كل من السدى والبطانة، فإن رأب القرنية الاختراقي (PKP) هو الخيار العملي. في المستقبل، تم اقتراح مفهوم “زراعة القرنية المتسلسلة” التي تجمع بين رأب القرنية السدوي العميق (DALK) لاستبدال السدى ورأب القرنية البطانية بغشاء ديسيميه (DMEK) لاستبدال البطانة على مراحل، ولكن كل هذه لا تزال في مرحلة البحث.
يُظهر تركيز الكيراتان سلفات في المصل انخفاضًا عن المستوى الطبيعي لدى مرضى حثل القرنية المخاطي (MCD) من النمط المناعي II، وتتم مناقشة فائدته كمؤشر أيضي جهازي 2). في المستقبل، من المتوقع استخدامه في الفحص عن طريق اختبارات الدم والتشخيص المبكر لحاملي الطفرات في عائلات CHST6. بالإضافة إلى ذلك، يمكن أن يساهم التحليل الآلي لصور المصباح الشقي باستخدام الذكاء الاصطناعي في الكشف المبكر عن الأمراض النادرة مثل هذا المرض.
نظرًا لأن المرض وراثي جسمي متنحي، فإن حوالي 25% من أشقاء المريض قد يصابون بالمرض. باستخدام اختبار جين CHST6، يمكن إجراء الفحص داخل الأسرة والاستشارة الوراثية فيما يتعلق بالزواج والحمل. يُوصى بالإحالة المبكرة إلى أخصائي في العائلات التي لديها تاريخ من زواج الأقارب أو حالات عتامة قرنية مماثلة داخل الأسرة.
Akama TO, Nishida K, Nakayama J, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nature Genetics. 2000;26(2):237-241. doi:10.1038/79987.
Aggarwal S, Peck T, Golen J, Karcioglu ZA. Macular corneal dystrophy: A review. Survey of Ophthalmology. 2018;63(5):609-617. doi:10.1016/j.survophthal.2018.03.004.
Jonasson F, Oshima E, Thonar EJ, et al. Macular corneal dystrophy in Iceland. A clinical, genealogic, and immunohistochemical study of 28 patients. Ophthalmology. 1996;103(7):1111-1117. doi:10.1016/s0161-6420(96)30559-9.
Jonasson F, Johannsson JH, Garner A, Rice NS. Macular corneal dystrophy in Iceland. Eye (London, England). 1989;3 ( Pt 4):446-54. doi:10.1038/eye.1989.66. PMID:2606219.
Sultana A, Sridhar MS, Jagannathan A, et al. Novel mutations of the carbohydrate sulfotransferase-6 (CHST6) gene causing macular corneal dystrophy in India. Mol Vis. 2003;9:730-734. PMID:14735064.
Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating keratoplasty for macular corneal dystrophy. Ophthalmology. 2005;112(2):220-224. doi:10.1016/j.ophtha.2004.08.017.
Reinhart WJ, Musch DC, Jacobs DS, Lee WB, Kaufman SC, Shtein RM. Deep anterior lamellar keratoplasty as an alternative to penetrating keratoplasty a report by the american academy of ophthalmology. Ophthalmology. 2011;118(1):209-218. doi:10.1016/j.ophtha.2010.11.002. PMID:21199711.
انسخ نص المقال والصقه في مساعد الذكاء الاصطناعي الذي تفضله.
تم نسخ المقال إلى الحافظة
افتح أحد مساعدي الذكاء الاصطناعي أدناه والصق النص المنسوخ في مربع المحادثة.