Distrofi kornea makular (MCD) adalah distrofi kornea herediter di mana glikosaminoglikan (terutama keratan sulfat) terakumulasi di stroma kornea. Penyakit ini mengikuti pola pewarisan resesif autosomal dan disebabkan oleh mutasi gen CHST6 yang terletak pada lengan panjang kromosom 16 (16q22)1,3). Dahulu disebut juga distrofi kornea Groenouw tipe II atau distrofi kornea Fehr.
Berbeda dengan banyak distrofi stroma kornea lainnya (granular, lattice) yang bersifat dominan autosomal, penyakit ini ditandai dengan pewarisan resesif autosomal. Di Jepang, penyakit ini dihitung sebagai salah satu dari empat distrofi kornea utama bersama dengan distrofi kornea granular (tipe I dan II), distrofi kornea lattice (tipe I dan IIIA), dan distrofi kornea gelatinous drop-like, yang bersama-sama mencakup sekitar 96% dari seluruh distrofi kornea. Dua yang pertama bersifat dominan autosomal, sedangkan dua yang terakhir (gelatinous drop-like dan macular) bersifat resesif autosomal.
Dalam klasifikasi IC3D (International Committee for Classification of Corneal Dystrophies), MCD diposisikan sebagai salah satu tipe distrofi stroma 1). Dalam tinjauan oleh Aggarwal dkk. di Survey of Ophthalmology 2018, penyakit ini dijelaskan sebagai “distrofi stroma yang jarang namun berdampak besar pada fungsi penglihatan”, dan sistem diagnosis serta tata laksana dirangkum 4). Dalam edisi ke-2 klasifikasi IC3D, distrofi kornea diklasifikasikan ke dalam kategori 1–4 berdasarkan kekuatan bukti gen penyebab, temuan patologis, dan gambaran klinis 1). MCD diklasifikasikan ke dalam kategori 1 (distrofi yang ditetapkan pada tingkat gen) karena identifikasi mutasi gen CHST6.
Secara historis, penyakit ini pertama kali dideskripsikan oleh Groenouw pada tahun 1890, kemudian muncul tradisi penamaan distrofi granular sebagai “tipe I” dan distrofi macular sebagai “tipe II”. Pada tahun 1938, Jones dan Zimmerman menetapkannya sebagai penyakit independen, dan pada tahun 2000, dasar molekuler dijelaskan oleh Akama dkk. melalui identifikasi gen CHST6 3).
Secara global, terdapat variasi geografis yang besar, dan di daerah dengan prevalensi tinggi, terdapat akumulasi dalam keluarga. Penyakit ini relatif jarang.
Variasi Geografis Prevalensi
Amerika Serikat: sekitar 0,3 per 250.000 orang, jarang 2,3)
Islandia: sekitar 19 per 250.000 orang, salah satu daerah dengan frekuensi tertinggi di dunia 5,6)
Daerah prevalensi tinggi: India Selatan, Arab Saudi, Islandia, dan Skandinavia 5,7)
Daerah lain: relatif jarang. Terjadi pada perkawinan sedarah atau heterozigot majemuk
Fenotipe Imun
Tipe I: negatif untuk keratan sulfat di kornea dan serum 2)
Tipe IA: positif di dalam keratosit kornea, negatif di serum 2)
Tipe II: positif untuk keratan sulfat di kornea dan serum 2)
Gambaran klinis: Fenotipe ketiga tipe identik dan tidak dapat dibedakan dengan slit lamp 2,8)
Fenotipe imun MCD diklasifikasikan berdasarkan jumlah keratan sulfat di kornea dan serum menggunakan antibodi monoklonal anti-keratan sulfat 2,3).
Fenotipe
Keratan sulfat kornea
Keratan sulfat serum
Tipe I
Negatif
Negatif
Tipe IA
Positif (intraseluler)
Negatif
Tipe II
Positif
Positif
Sebagian besar pasien tergolong tipe I atau IA. Namun secara klinis, perbedaan subtipe ini tidak penting dan tidak dapat dibedakan melalui pemeriksaan2,8).
QApa perbedaan distrofi kornea makular dengan distrofi kornea lainnya?
A
Perbedaan terbesar adalah pola pewarisan autosomal resesif. Distrofi kornea granular dan kisi bersifat autosomal dominan, sedangkan penyakit ini memerlukan mutasi pada kedua alel gen CHST6. Selain itu, ditandai dengan kekeruhan difus seperti kaca buram, seluruh kornea menjadi putih, dan pada pemeriksaan slit-lamp, terdapat deposit di lapisan superfisial di bagian tengah dan di lapisan dalam di perifer.
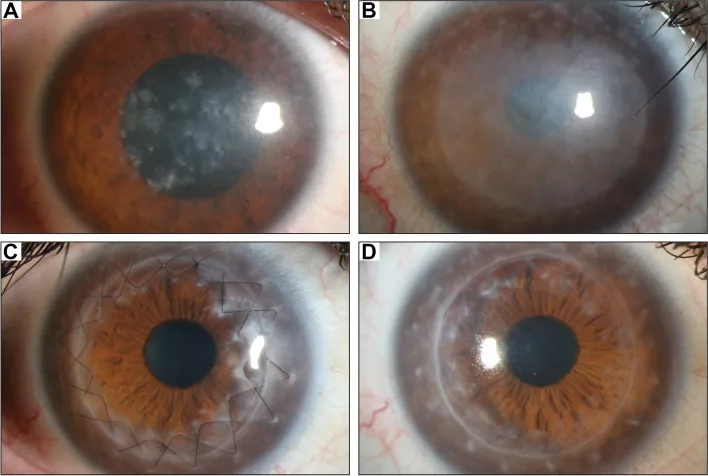
Gassel CJ, et al. Histological findings of corneal tissue after failed phototherapeutic keratectomy in macular corneal dystrophy - a case report. BMC Ophthalmol. 2022. Figure 1. PMCID: PMC9080147. License: CC BY.
Foto slit-lamp menunjukkan kekeruhan difus putih keabu-abuan dan deposit makula dari pusat hingga seluruh kornea. Menunjukkan temuan klinis khas distrofi kornea makular, memudahkan pemahaman tentang kekeruhan kornea yang menyebabkan penurunan penglihatan.
Pola progresi khas temuan klinis adalah sebagai berikut.
Secara klinis, deposit halus difus terlihat di stroma kornea, menyebabkan kekeruhan seperti kaca buram. Seiring progresi, kekeruhan meluas ke seluruh ketebalan stroma, dan menyebar dari pusat ke perifer. Kemudian, selain kekeruhan ringan, banyak kekeruhan kecil tidak beraturan berwarna putih keabu-abuan terlihat di stroma superfisial hingga dalam.9)
Temuan awal
Kekeruhan bercak: Kekeruhan bercak kecil putih keabu-abuan muncul di stroma superfisial kornea sentral
Kekeruhan seperti kaca buram: Kekeruhan ringan difus terlihat di stroma kornea
Batas tidak jelas: Tepi kekeruhan tidak tajam, batas dengan stroma normal tidak jelas
Tanda Stadium Lanjut
Perluasan ke seluruh lapisan: Kekeruhan meluas ke seluruh lapisan stroma
Perluasan ke perifer: Kekeruhan menyebar dari pusat ke perifer
Endapan pada endotel dan membran Descemet: Zat abnormal juga terakumulasi di struktur dalam
Pada slit-lamp, seluruh kornea tampak keruh difus dengan endapan plak abu-abu keputihan yang tidak teratur. Dengan membuat irisan optik menggunakan celah cahaya, terlihat distribusi khas di mana endapan terletak di lapisan superfisial di bagian sentral dan di lapisan dalam di bagian perifer. Lesi bercak sering muncul dalam pola melingkar konsentris8).
Kekeruhan dapat meluas hingga ke limbus, dan ini merupakan poin diferensial penting dari distrofi kornea lainnya. Pada distrofi kornea granular dan distrofi kornea lattice, limbus sering tetap jernih, sedangkan pada MCD, seluruh kornea sering keruh hingga ke limbus2,8). Selain itu, munculnya astigmatisme ireguler terkait dengan endapan di stroma anterior, dan dapat terjadi penurunan sensasi kornea. Karena endapan zat abnormal juga terjadi pada endotel, pada kasus lanjut dapat terjadi edema stroma akibat penurunan fungsi endotel8).
Perjalanan alami bervariasi antar individu, tetapi sering melalui tahap-tahap berikut.
Masa bayi dan anak-anak (fase asimtomatik): Mutasi gen sudah ada sejak lahir, tetapi temuan slit-lamp sedikit dan tidak bergejala
Usia sekolah hingga remaja (fase kekeruhan awal): Kekeruhan difus ringan muncul di lapisan superfisial stroma kornea, dan kemudian endapan bercak terlihat
10-30 tahun (fase penurunan penglihatan): Kekeruhan berkembang, dan pasien mulai menyadari penurunan penglihatan
30-40 tahun (fase lanjut): Kekeruhan meluas ke seluruh lapisan stroma dan limbus, penurunan sensasi kornea, penipisan kornea, dan astigmatisme ireguler menjadi jelas
Usia paruh baya ke atas (fase indikasi transplantasi kornea): Penurunan fungsi penglihatan mencapai tingkat yang mengganggu aktivitas sehari-hari, dan transplantasi dipertimbangkan
MCD adalah penyakit progresif di mana penurunan fungsi penglihatan berlanjut sepanjang hidup, berbeda dengan distrofi kornea granular tipe I dan beberapa subtipe distrofi kornea lattice yang gangguan penglihatannya ringan4,8).
Gen penyebabnya adalah CHST6 (carbohydrate sulfotransferase 6)3). Terletak pada kromosom 16q22 dan mengkode enzim yang mentransfer gugus sulfat ke N-asetilglukosamin pada proteoglikan kornea. Jenis mutasi gen ini sangat beragam, termasuk mutasi missense, nonsense, frameshift, dan delesi pada daerah hulu 5’, yang telah dilaporkan pada berbagai kelompok etnis 3,7).
Karena pewarisan bersifat autosomal resesif, biasanya kedua orang tua dari probandus adalah pembawa. Frekuensi kejadian cenderung lebih tinggi di daerah atau kelompok dengan banyak perkawinan sedarah. Penyakit juga dapat terjadi pada heterozigot majemuk yang lahir dari perkawinan antar keluarga yang berbeda 7).
Akama dkk. pada tahun 2000 mengidentifikasi CHST6 sebagai gen penyebab penyakit ini, dan menunjukkan bahwa baik fenotipe imun tipe I maupun tipe II disebabkan oleh mutasi pada lokus gen yang sama 3). Temuan ini merupakan penemuan penting yang menunjukkan bahwa perbedaan fenotipe imun ditentukan oleh pola mutasi yang berbeda pada satu gen, dan menjadi dasar sistem diagnosis genetik selanjutnya.
Lebih dari 200 jenis mutasi telah dilaporkan, dengan mutasi missense yang paling umum. Sultana dkk. mengidentifikasi banyak mutasi baru pada kelompok pasien di India Selatan, dan menunjukkan bahwa tingginya frekuensi di wilayah tersebut disebabkan oleh akumulasi mutasi spesifik lokal 7). Akumulasi regional serupa juga dilaporkan di Arab Saudi dan Islandia, dan diyakini bahwa latar belakang sejarah populasi (efek pendiri dan kebiasaan perkawinan sedarah) berkontribusi pada frekuensi kejadian 5,6,7).
Frekuensi kejadian bervariasi menurut wilayah, dan penyakit ini relatif jarang 9). Dibandingkan dengan distrofi kornea lainnya seperti distrofi kornea granular, jumlah kasus lebih sedikit, dan cenderung dilaporkan pada keluarga dengan latar belakang perkawinan sedarah atau heterozigot majemuk.
Riwayat keluarga: Karena pewarisan autosomal resesif, kedua orang tua harus merupakan pembawa
Perkawinan sedarah: Meningkatkan angka kejadian
Faktor geografis: Prevalensi tinggi di India Selatan, Arab Saudi, Islandia, dan Skandinavia 5,7)
QApakah tes genetik diperlukan?
A
Tes gen CHST6 berguna untuk diagnosis pasti. Tes genetik dapat dilakukan di fasilitas medis yang terakreditasi. Namun, di banyak fasilitas, diagnosis klinis terutama didasarkan pada pemeriksaan slit-lamp. Tes genetik berguna untuk menilai risiko pada anak di masa depan, dan untuk memastikan diagnosis pada kasus dengan gambaran klinis yang tidak khas.
Kecurigaan MCD muncul pada pasien remaja hingga dewasa muda dengan kekeruhan difus seluruh kornea yang bilateral dan progresif. Pertama, anamnesis rinci tentang gejala subjektif (penurunan visus, fotofobia, iritasi), riwayat keluarga, dan perkawinan sedarah. Selanjutnya, evaluasi temuan kornea dengan slit-lamp, penilaian fungsi endotel, dan jika perlu, tes genetik.
Ini adalah pemeriksaan dasar untuk diagnosis. Jika ditemukan kekeruhan kornea bilateral tanpa injeksi atau edema, curigai distrofi kornea. Pada MCD, temuan berikut khas:
Kekeruhan difus seperti kaca buram: Seluruh kornea keruh secara difus
Deposit bercak: Banyak kekeruhan tidak teratur berwarna putih keabu-abuan
Distribusi konsentris: Pada potongan cahaya celah, lapisan superfisial di sentral dan lapisan dalam di perifer
Infiltrasi limbus: Kekeruhan dapat meluas hingga ke limbus
Kelainan permukaan endotel: Pada kasus lanjut, dapat ditemukan deposit gutata
Kebanyakan distrofi kornea diamati pada level celah sebagai lesi diskontinu (dengan area jernih di antara deposit), sedangkan MCD secara luar biasa menunjukkan pola kekeruhan difus. Bersama dengan distrofi kornea lattice tipe I dan distrofi kornea tetesan gelatin, ini merupakan contoh khas dari “deposit kornea yang diamati secara difus”.
Optical Coherence Tomography Segmen Anterior (AS-OCT): Memvisualisasikan distribusi deposit di lapisan superfisial dan dalam kornea
Mikroskopi Konfokal (in vivo confocal microscopy): Menunjukkan material reflektif tinggi dengan batas tidak jelas dan hilangnya gambaran keratosit normal8)
Topografi Kornea: Menunjukkan peningkatan densitas puncak kornea dan penipisan kornea sentral
Mikroskop Ultrasonik Biologis (UBM): Berguna untuk menilai kekeruhan dalam dan struktur posterior kornea
Mikroskop Specular: Evaluasi kepadatan dan morfologi sel endotel. Tingkat lesi endotel berhubungan langsung dengan pilihan teknik bedah
Pentacam Kornea (Pencitraan Scheimpflug): Memberikan peta densitas seluruh lapisan kornea, berguna untuk evaluasi tiga dimensi area kekeruhan
Secara histologis, pewarnaan Alcian blue dan pewarnaan besi koloid menunjukkan hasil positif, dan terlihat akumulasi difus glikosaminoglikan sulfat rendah di dalam dan di luar sel keratosit stroma kornea2,8). Robekan membran Bowman dapat diamati, dan pada kasus lanjut, zat abnormal juga ditemukan di dalam sel endotel. Temuan seperti guttae juga dapat muncul pada membran Descemet.
Selain itu, distrofi kornea posterior amorf (PACD) dan distrofi kornea pre-Descemet (PDCD) juga termasuk dalam diagnosis banding. Mukopolisakaridosis sistemik (seperti sindrom Hurler, Scheie, Morquio) juga dapat menyebabkan kekeruhan kornea, sehingga perlu dievaluasi termasuk temuan sistemik8).
QBagaimana distrofi kornea makula didiagnosis?
A
Diagnosis klinis ditegakkan dengan slit lamp yang menunjukkan kekeruhan kornea difus seperti kaca buram dan deposit makula, serta bilateral, progresif, riwayat keluarga, dan usia onset (10-30 tahun). Tes genetik CHST6 berguna untuk konfirmasi.
Tujuan perawatan MCD adalah: (1) mempertahankan atau memulihkan fungsi penglihatan, (2) mengurangi nyeri dan gejala iritasi dengan menstabilkan permukaan mata, (3) mencegah komplikasi (erosi epitel dan infeksi). Karena tidak ada pengobatan kausal yang mendasar, intervensi dilakukan secara bertahap sesuai stadium dan gejala.
Pada kasus dengan penurunan penglihatan yang progresif, transplantasi kornea adalah satu-satunya pengobatan kuratif. Teknik dipilih berdasarkan ada tidaknya penyakit endotel.
Keroplasti lamela anterior dalam (DALK): Merupakan pilihan pertama pada kasus tanpa keterlibatan endotel 8,9). Karena mempertahankan endotel kornea sendiri, risiko penolakan cangkok lebih rendah dibandingkan keroplasti penetrasi penuh. Dalam laporan evaluasi AAO (American Academy of Ophthalmology), DALK untuk distrofi stroma dinilai memberikan pemulihan fungsi penglihatan yang setara dengan PKP dengan kehilangan endotel yang lebih sedikit 8). Tingkat rekurensi setelah DALK rendah, dan rekurensi jarang terjadi karena stroma resipien diganti.
Keroplasti penetrasi penuh (PKP): Diindikasikan pada kasus lanjut dengan keterlibatan endotel atau membran Descemet dengan deposit zat abnormal, serta kasus dengan penipisan kornea sentral yang parah 7). Usia rata-rata PKP pertama untuk MCD adalah 30-40 tahun 7), dan tingkat kelangsungan hidup cangkok baik.
Keratektomi fototerapeutik (PTK): Dilakukan secara simtomatik untuk erosi epitel berulang atau kekeruhan sikatrik superfisial. Namun, perlu diperhatikan hiperopia dan induksi kekeruhan stroma.
Pada MCD, zat abnormal juga dapat mengendap di sel endotel, sehingga pada kasus dengan penyakit yang meluas ke dalam, PKP cenderung dipilih daripada DALK7). Jika terdapat kelainan endotel, keroplasti penetrasi penuh diindikasikan. Evaluasi endotel praoperasi yang terperinci (mikroskop spekular atau konfokal) menjadi kunci dalam menentukan teknik bedah.
Tingkat kekambuhan setelah DALK dilaporkan rendah, yaitu sekitar 5%. Sementara itu, tingkat kelangsungan hidup cangkok setelah PKP baik, dan banyak laporan menunjukkan kelangsungan hidup jangka panjang, namun beberapa kasus melaporkan pengendapan kembali zat abnormal pada cangkok setelah beberapa tahun hingga belasan tahun pasca operasi8). Dalam seri Al-Swailem dkk. di Arab Saudi, tingkat kelangsungan hidup cangkok setelah PKP untuk MCD baik, tetapi kekambuhan diamati pada beberapa kasus selama follow-up jangka panjang8). Selain itu, dalam tinjauan AAO oleh Reinhart dkk., ditunjukkan bahwa DALK mencapai pemulihan visual yang setara atau lebih baik daripada PKP dengan tingkat kehilangan sel endotel yang rendah9). Studi multisenter Unal dkk. juga melaporkan efektivitas DALK pada distrofi stroma termasuk MCD.
Secara umum, kekambuhan pasca transplantasi kornea terjadi karena patologi molekuler penyakit asli masih ada di pihak inang. Pada DALK, endotel inang dan lapisan anterior membran Descemet dipertahankan, sehingga perlu diperhatikan bahwa penyakit dapat berkembang pada kasus dengan lesi endotel. Setelah operasi, diperlukan pemantauan berkala jangka panjang (pemeriksaan visus, slit lamp, pengukuran kepadatan sel endotel).
QManakah yang harus dipilih, DALK atau PKP?
A
Jika endotel atau membran Descemet tidak terlibat, transplantasi lapisan dalam anterior dalam (DALK) adalah pilihan pertama. DALK mempertahankan endotel kornea sendiri, sehingga risiko penolakan rendah, dan tingkat kekambuhan dilaporkan sekitar 5% setelah operasi. Di sisi lain, pada kasus di mana terdapat deposit zat abnormal juga di endotel, transplantasi kornea lapisan penuh (PKP) diindikasikan. Teknik operasi ditentukan berdasarkan evaluasi endotel praoperasi.
6. Fisiopatologi dan Mekanisme Terjadinya Secara Detail
Gen CHST6 mengkode karbohidrat sulfotransferase 6 (carbohydrate sulfotransferase 6)3). Enzim ini bertanggung jawab untuk transfer gugus sulfat ke N-asetilglukosamin pada molekul keratan, dan penting untuk sintesis normal keratan sulfat (KS) yang terkandung dalam proteoglikan kornea.
Ketika aktivitas enzim hilang karena mutasi gen, keratan sulfat yang tersulfasi rendah dan tidak sempurna disintesis. Keratan sulfat abnormal ini memiliki kelarutan rendah dan mengendap secara abnormal di dalam dan di luar sel keratosit di stroma kornea2,3).
Kelainan kuantitatif dan kualitatif keratan sulfat menyebabkan rangkaian patologis berikut.
Produksi proteoglikan kecil kaya leusin (SLRP) yang abnormal: SLRP khas kornea seperti lumican, keratocan, dan mimecan tidak disintesis secara normal
Kelainan susunan serat kolagen: SLRP ini secara ketat mengontrol diameter serat kolagen kornea dan jarak antar serat, menjamin transparansi. Penurunan fungsi SLRP menyebabkan diameter serat kolagen menjadi tidak seragam dan jarak antar serat berubah 2)
Akumulasi abnormal di matriks ekstraseluler: Keratan yang tidak tersulfatasi sendiri mengendap di matriks ekstraseluler
Peningkatan hamburan cahaya dan hilangnya transparansi: Perubahan gabungan di atas meningkatkan hamburan cahaya tampak, menyebabkan kekeruhan difus di seluruh kornea
Akumulasi glikosaminoglikan diamati di dalam dan di luar sel keratosit stroma, dan seiring perkembangan penyakit, menyebar ke membran Bowman, membran Descemet, dan sel endotel. Pada tipe I, penurunan aktivitas enzim juga telah dikonfirmasi di kartilago aurikuler, menunjukkan kemungkinan bahwa ini adalah bagian dari gangguan metabolisme keratan sulfat sistemik 2). Namun, gejala sistemik jarang muncul secara klinis, dan kondisi ini diperlakukan sebagai penyakit lokal dengan gejala utama di kornea.
Di antara proteoglikan kornea, lumican berperan mengontrol diameter serat kolagen hingga sekitar 25 nm, sedangkan keratocan dan mimecan menjaga jarak antar serat tetap seragam. Ketika rantai samping tersulfatasi dari SLRP ini pendek dan tidak lengkap, serat kolagen bervariasi dalam ketebalan dan jarak antar serat menjadi tidak seragam. Akibatnya, hamburan cahaya di dalam stroma kornea meningkat, dan secara klinis diamati sebagai kekeruhan seperti kaca buram.
Teori kisi Maurer secara klasik dikenal untuk menjaga transparansi kornea melalui “pembatalan interferensi cahaya oleh susunan kisi serat kolagen yang teratur”, tetapi pada MCD, struktur kisi ini rusak karena kelainan SLRP, sehingga transparansi hilang 2,4).
Dalam penelitian dasar terbaru, dilaporkan bahwa disfungsi autofagi (self-eating) yang disebabkan oleh mutasi CHST6 dapat menginduksi piroptosis (kematian sel inflamasi) keratosit, berkontribusi pada perkembangan penyakit 8). Kelainan autofagi serupa juga telah dilaporkan pada distrofi kornea lainnya (seperti tipe granular II), menarik perhatian sebagai patomekanisme umum distrofi kornea secara keseluruhan.
Terapi gen yang ditargetkan telah diusulkan sebagai strategi pengobatan permanen 8). Penelitian dasar tentang penyuntingan gen menggunakan CRISPR/Cas9 sedang berlangsung pada distrofi epitel kornea Meesmann, dan berpotensi menjadi pilihan pengobatan di masa depan untuk MCD. Namun, masih banyak tantangan untuk aplikasi klinis, seperti penyuntingan alel normal yang tidak disengaja (efek di luar target), pengembangan metode penghantaran gen yang efisien ke sel stroma kornea, dan verifikasi keamanan jangka panjang.
Pendekatan untuk menghilangkan keratan sulfat yang kurang tersulfasi yang terakumulasi di kornea secara enzimatik sedang diteliti secara dasar. Ini menerapkan konsep terapi penggantian enzim yang digunakan pada mukopolisakaridosis sistemik secara lokal ke kornea, tetapi belum ada laporan aplikasi klinis saat ini.
Descemet stripping automated endothelial keratoplasty (DSAEK) dan Descemet membrane endothelial keratoplasty (DMEK) adalah teknik yang dikembangkan terutama untuk distrofi endotel Fuchs dan keratopati bulosa, tetapi penerapannya pada kasus MCD dengan keterlibatan endotel awal masih menjadi subjek penelitian di masa depan. Saat ini, karena penyakit ini mempengaruhi baik stroma maupun endotel, keratoplasti penetrasi (PKP) adalah pilihan yang realistis. Di masa depan, konsep “keratoplasti sekuensial” yang menggabungkan deep anterior lamellar keratoplasty (DALK) untuk mengganti stroma dan DMEK untuk mengganti endotel secara bertahap telah diusulkan, tetapi semuanya masih dalam tahap penelitian.
Kadar keratan sulfat serum dilaporkan lebih rendah dari normal pada pasien MCD dengan fenotip imun tipe II, dan kegunaannya sebagai penanda metabolik sistemik sedang dibahas 2). Di masa depan, diharapkan dapat digunakan untuk skrining melalui tes darah dan diagnosis dini pembawa mutasi pada keluarga CHST6. Selain itu, analisis otomatis gambar slit-lamp menggunakan kecerdasan buatan juga dapat berkontribusi pada deteksi dini penyakit langka seperti ini.
Karena penyakit ini bersifat resesif autosomal, sekitar 25% saudara kandung pasien dapat terkena. Dengan menggunakan tes gen CHST6, skrining keluarga dan konseling genetik mengenai pernikahan dan kehamilan dapat dilakukan. Rujukan dini ke spesialis dianjurkan pada keluarga dengan riwayat perkawinan sedarah atau kasus kekeruhan kornea serupa dalam keluarga.
Akama TO, Nishida K, Nakayama J, et al. Macular corneal dystrophy type I and type II are caused by distinct mutations in a new sulphotransferase gene. Nature Genetics. 2000;26(2):237-241. doi:10.1038/79987.
Aggarwal S, Peck T, Golen J, Karcioglu ZA. Macular corneal dystrophy: A review. Survey of Ophthalmology. 2018;63(5):609-617. doi:10.1016/j.survophthal.2018.03.004.
Jonasson F, Oshima E, Thonar EJ, et al. Macular corneal dystrophy in Iceland. A clinical, genealogic, and immunohistochemical study of 28 patients. Ophthalmology. 1996;103(7):1111-1117. doi:10.1016/s0161-6420(96)30559-9.
Jonasson F, Johannsson JH, Garner A, Rice NS. Macular corneal dystrophy in Iceland. Eye (London, England). 1989;3 ( Pt 4):446-54. doi:10.1038/eye.1989.66. PMID:2606219.
Sultana A, Sridhar MS, Jagannathan A, et al. Novel mutations of the carbohydrate sulfotransferase-6 (CHST6) gene causing macular corneal dystrophy in India. Mol Vis. 2003;9:730-734. PMID:14735064.
Al-Swailem SA, Al-Rajhi AA, Wagoner MD. Penetrating keratoplasty for macular corneal dystrophy. Ophthalmology. 2005;112(2):220-224. doi:10.1016/j.ophtha.2004.08.017.
Reinhart WJ, Musch DC, Jacobs DS, Lee WB, Kaufman SC, Shtein RM. Deep anterior lamellar keratoplasty as an alternative to penetrating keratoplasty a report by the american academy of ophthalmology. Ophthalmology. 2011;118(1):209-218. doi:10.1016/j.ophtha.2010.11.002. PMID:21199711.
Salin teks artikel dan tempelkan ke asisten AI pilihan Anda.
Artikel disalin ke papan klip
Buka asisten AI di bawah, lalu tempelkan teks yang disalin ke kotak chat.