Distrofi kornea kisi (lattice corneal dystrophy, LCD) adalah distrofi kornea herediter dengan deposisi amiloid di stroma kornea yang menyebabkan kekeruhan linier seperti kisi. Penyakit ini pertama kali dideskripsikan pada tahun 1890-an, dan dalam klasifikasi IC3D edisi ke-2, diklasifikasikan menjadi LCD1 dan variannya (tipe 3, 3A, 1/3A, 4 sebelumnya)4).
LCD1, distrofi kornea granular, distrofi Reis-Bücklers, dan distrofi Thiel-Behnke membentuk kelompok penyakit yang disebut “distrofi terkait TGFBI”. Gen penyebab TGFBI (transforming growth factor beta-induced gene) terletak pada lengan panjang kromosom 5 (5q31) dan mengikuti pola pewarisan autosomal dominan. Protein TGFBI (TGFBIp, kerato-epithelin, βig-h3) diproduksi oleh epitel kornea dan didistribusikan ke seluruh lapisan kornea. Di stroma kornea, protein ini berperan dalam pembentukan serat kolagen. Meskipun mutasi terjadi pada gen yang sama, perbedaan lokasi mutasi dan asam amino pengganti menyebabkan perbedaan besar dalam substansi yang terdeposisi (hialin atau amiloid) dan gambaran klinis5).
Mutasi representatif LCD1 adalah R124C, yaitu substitusi arginin pada posisi 124 gen TGFBI menjadi sistein. Pada varian LCD IIIA, telah dilaporkan mutasi seperti L527R.
Protein abnormal yang terakumulasi di kornea menunjukkan warna merah dengan pewarnaan Congo red, dan menunjukkan birefringensi hijau apel yang khas di bawah mikroskop polarisasi, sehingga dipastikan sebagai amiloid. Temuan ini merupakan indikator diagnosis jaringan klasik untuk amiloidosis sejak abad ke-196).
Jenis yang sebelumnya disebut “distrofi kisi kornea tipe 2” adalah manifestasi okular dari amiloidosis sistemik tipe gelsolin (GSN-AMYL, sindrom Meretoja), dan dalam klasifikasi IC3D saat ini diklasifikasikan sebagai “amiloidosis familial” dan ditangani secara independen dari LCD klasik4,10). Sindrom ini pertama kali dijelaskan oleh Meretoja di Finlandia pada tahun 1969, merupakan penyakit herediter yang ditandai dengan kekeruhan kisi kornea disertai neuropati kranial progresif, kulit kendur, dan gejala sistemik10,11). Karena pentingnya membedakan keduanya dalam praktik klinis, kedua kondisi tersebut disertakan dalam artikel ini.
Kekeruhan filamen kontur ganda di area pupil, erosi epitel berulang
LCD IIIA (tipe varian)
TGFBI (5q31)
L527R dll.
Setelah usia 40 tahun
Garis kisi tebal seperti tali di stroma dalam, tidak ada erosi epitel
Tipe GSN (Meretoja)
GSN (9q34)
D187N, p.Glu580Lys2)
30-40 tahun
Garis kisi radial di perifer, amiloidosis sistemik
Di Jepang, distrofi terkait TGFBI yang paling sering adalah tipe granular II (tipe Avellino, R124H), dan LCD1 lebih jarang dibandingkan. Namun, karena keduanya hanya berbeda beberapa basa pada gen TGFBI yang sama, pemeriksaan genetik dianjurkan untuk konfirmasi pada kasus dengan gambaran klinis yang tumpang tindih. Prevalensi pasti LCD secara keseluruhan di Jepang belum dilaporkan, tetapi termasuk relatif jarang di antara distrofi kornea.
QApa perbedaan antara LCD1 dan sindrom Meretoja?
A
LCD1 adalah deposit amiloid terbatas pada kornea akibat mutasi gen TGFBI, timbul dari area pupil pada usia 10-20 tahun dan sering disertai erosi epitel berulang. Sebaliknya, sindrom Meretoja (sebelumnya LCD2, tipe GSN) adalah manifestasi okular dari amiloidosis sistemik akibat mutasi gen GSN (gelsolin), timbul dari perifer kornea pada usia 30-40 tahun, dan transparansi sentral bertahan lama. Sindrom Meretoja disertai gejala sistemik seperti kulit kendur, wajah seperti topeng, neuropati perifer, dan aritmia jantung2,10). Dalam IC3D edisi kedua, sindrom Meretoja diklasifikasikan terpisah dari distrofi kornea lattice4).
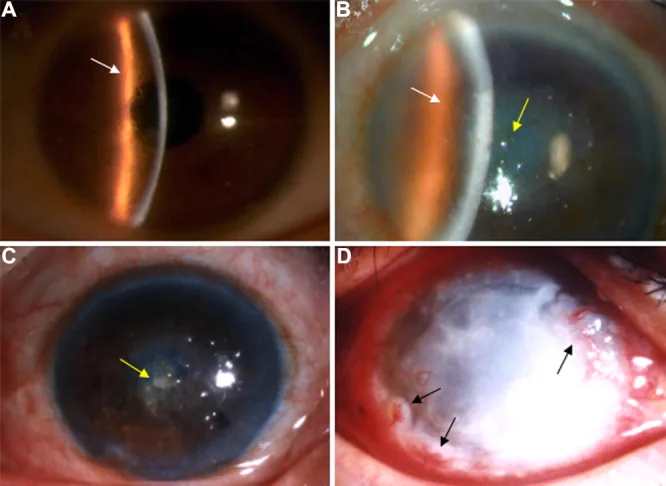
Liu Z, et al. An R124C mutation in TGFBI caused lattice corneal dystrophy type I with a variable phenotype in three Chinese families. Mol Vis. 2008. Figure 2. PMCID: PMC2443752. License: CC BY.
Pada foto slit-lamp, terlihat garis-garis bercabang seperti kisi di stroma kornea dan kekeruhan dominan di bagian tengah. Gambaran ini menunjukkan temuan klinis khas dari distrofi kornea kisi.
Pada LCD1, sebagian besar pasien tidak bergejala pada masa kanak-kanak, hanya ditemukan kekeruhan halus yang baru terdeteksi dengan iluminasi retro dari slit-lamp. Setelah usia 10-20 tahun, terjadi erosi epitel kornea berulang (RCE) secara berulang, dengan nyeri mata hebat saat bangun tidur, fotofobia, lakrimasi, dan sensasi benda asing. Sekitar usia 30 tahun, muncul kekeruhan putih di lapisan superfisial stroma kornea sentral, dan penurunan penglihatan progresif setelah usia 40 tahun.
Pada LCD IIIA (tipe varian), kerusakan epitel biasanya tidak terjadi, dan keluhan utama adalah penurunan penglihatan lambat setelah usia 40 tahun.
Pada LCD2 lama (sindrom Meretoja), gejala mata muncul pada usia 30-40 tahun, tetapi gangguan penglihatan berat sering tertunda hingga usia 60 tahun11). Gejala sistemik seperti kulit kelopak mata kendur, wajah seperti topeng, neuropati kranial progresif, dan aritmia jantung sering mendahului atau menyertai2,10).
Berikut adalah temuan slit-lamp untuk setiap tipe penyakit.
LCD1 (tipe klasik)
Lokasi awal: Muncul sebagai kekeruhan titik-titik halus atau garis di area pupil kedua mata, dari lapisan Bowman hingga stroma superfisial.
Garis kisi: Kekeruhan seperti benang atau garis dengan kontur ganda yang saling bertaut membentuk kekeruhan seperti jaring atau bintang.
Tahap lanjut: Muncul kekeruhan putih susu berbentuk oval atau bulat di kornea sentral.
Iluminasi retro: Garis kisi tipis tembus pandang yang sulit dilihat dengan iluminasi langsung menjadi jelas terlihat.
Pewarnaan fluorescein: Karena adhesi epitel yang buruk, permukaan kornea menjadi kasar.
Erosi epitel berulang: Sering terjadi karena deposit meluas ke sel basal epitel dan membran Bowman.
LCD IIIA (tipe varian)
Garis kisi: Garis kisi tebal dan panjang di lapisan tengah hingga dalam stroma, kadang bercabang seperti pohon. Dapat diamati bahkan dengan iluminasi langsung.
Fenotipe: Ada tiga pola: ① hanya garis kisi, ② hanya endapan granular kecil, ③ campuran keduanya. Pada individu yang sama, mata kanan dan kiri dapat menunjukkan fenotipe berbeda, dan kasus unilateral juga ada.
Epitel: Biasanya tidak terjadi gangguan epitel.
Homozigot: Pada homozigot L527R, garis kisi lebih tebal dan endapan granular sentral lebih besar, namun perbedaannya tidak setajam antara heterozigot dan homozigot R124H (tipe granular II).
Tipe GSN (Meretoja)
Garis kisi: Sejumlah kecil endapan kisi yang kurang halus muncul secara radial dari perifer.
Transparansi sentral: Area sentral tetap transparan untuk waktu yang lama setelah onset.
Erosi epitel: Jarang.
Temuan sistemik: Perubahan wajah seperti wajah topeng, bibir menonjol dengan gangguan gerakan, telinga terkulai, dan kelopak mata kendur2).
Pada LCD1, kekeruhan sirkular sentral dapat menjadi sangat parah pada beberapa kasus; telah dilaporkan pasien berusia 56 tahun dengan heterozigot R124C yang menjalani transplantasi kornea karena kekeruhan sirkular sentral.
QBisakah LCD1 didiagnosis pada anak-anak?
A
Sebagian besar LCD1 pada masa kanak-kanak tidak bergejala, dan kelainan sulit ditemukan dengan iluminasi langsung saja. Dengan pemeriksaan detail menggunakan slit lamp dengan teknik retroiluminasi atau iluminasi pantulan, dapat diamati kekeruhan titik-titik halus hingga linier di lapisan superfisial stroma sentral. Pada anak dengan erosi epitel kornea berulang, disarankan untuk mempertimbangkan LCD1, termasuk anamnesis riwayat keluarga dan pemeriksaan kornea orang tua. Tes gen TGFBI berguna untuk diagnosis pasti.
Mutasi khas LCD1: R124C (Arg124Cys) adalah yang paling sering5).
Mutasi khas LCD IIIA: Telah dilaporkan mutasi seperti L527R (Leu527Arg). Terdapat pula kasus homozigot.
Mutasi de novo: Telah dilaporkan kasus mutasi de novo L509P pada TGFBI yang menunjukkan fenotipe LCD IIIA1). Mutasi tidak ditemukan pada orang tua, dan diwariskan kepada salah satu anak1).
Peran TGFBIp: Diproduksi oleh epitel kornea dan tersebar di seluruh lapisan kornea, berperan dalam pembangunan serat kolagen di stroma5).
Terkait GSN (Sindrom Meretoja, sebelumnya LCD2)
Lokus genetik: 9q34 (gen GSN, gelsolin).
Pola pewarisan: Autosomal dominan.
Mutasi klasik: D187N (tipe Finlandia) adalah yang paling umum, dan p.Asp187Tyr juga telah dilaporkan10,11).
Mutasi baru: p.Glu580Lys yang dilaporkan pada keluarga Slovenia terletak di batas domain G4-G5, menyebabkan tolakan elektrostatik karena substitusi muatan negatif menjadi positif2).
Gambaran klinis: Selain kekeruhan kornea seperti kisi, terdapat amiloidosis sistemik dengan kulit kendur, aritmia jantung, gangguan ginjal, dan neuropati optik2).
Karena penyakit ini bersifat herediter, riwayat keluarga merupakan faktor risiko terpenting. Namun, mutasi de novo pada TGFBI dapat terjadi, sehingga tidak adanya riwayat keluarga tidak dapat menyingkirkan penyakit1). Pola pewarisan adalah autosomal dominan, dan jika salah satu orang tua adalah pembawa mutasi, kemungkinan diturunkan ke anak adalah 50%. Tidak ada perbedaan jenis kelamin, dan perbedaan ras tidak jelas pada LCD1, namun Sindrom Meretoja diketahui terkumpul pada keluarga di Finlandia11).
Kontribusi faktor lingkungan tidak jelas, dan onset serta perkembangan penyakit pada dasarnya ditentukan oleh genotipe. Namun, frekuensi erosi epitel berulang dapat memburuk pada lingkungan kering, penggunaan lensa kontak, atau trauma. Operasi refraktif (seperti LASIK, SMILE) dapat menyebabkan perburukan cepat distrofi kornea terkait TGFBI, sehingga diperlukan kewaspadaan pada kasus dengan riwayat keluarga saat skrining praoperasi5).
Untuk membedakan LCD1, varian, dan tipe GSN, diperlukan kombinasi temuan slit-lamp, histologi, dan genetik.
Pemeriksaan Klinis
Mikroskop Slit-Lamp: Pada pencahayaan langsung, garis kisi awal mudah terlewatkan. Dengan metode transiluminasi, kekeruhan halus dengan latar belakang pupil terdeteksi, dan dengan metode iluminasi retro, garis kisi tipis tembus pandang terdeteksi.
Pewarnaan Fluoresein: Pada LCD1, adhesi epitel menurun sehingga pewarnaan tampak kasar. Juga berguna untuk menilai luasnya erosi epitel.
OCT Segmen Anterior: Dapat mengukur kedalaman deposit secara berlapis. Pengukuran kedalaman lesi dengan FD-OCT berguna untuk menentukan kedalaman eksisi pada PTK1).
Mikroskop KonfokalKornea: Deposit di stroma dapat diamati pada tingkat seluler.
Diagnosis Pasti
Tes Genetik: Deteksi mutasi pada gen TGFBI dan GSN memastikan tipe penyakit. Meskipun fenotip sama, kecepatan rekurensi dan progresi berbeda tergantung mutasi, sehingga berdampak langsung pada rencana pengobatan.
Pemeriksaan Patologi: Pewarnaan Congo Red menunjukkan warna merah, dan di bawah mikroskop polarisasi menunjukkan birefringensi hijau apel, memastikan adanya amiloid6).
Imunohistokimia: Dapat membedakan tipe penyakit menggunakan antibodi anti-TGFBIp dan antibodi anti-gelsolin.
Anamnesis Riwayat Keluarga: Karena pewarisan autosomal dominan, pemeriksaan kornea orang tua dan saudara kandung mendukung diagnosis.
Distrofi Kornea Granular Tipe II (Tipe Avellino, TGFBI R124H): Merupakan distrofi terkait TGFBI paling umum di Jepang, menunjukkan campuran deposit granular dan garis kisi. Untuk diagnosis banding dengan LCD1, tes genetik dapat diandalkan.
Amiloidosis kornea sekunder: Tidak bersifat herediter, amiloid mengendap secara sekunder akibat stimulasi kronis permukaan mata seperti trikiasis atau keratokonus. Perbedaan utamanya adalah tidak adanya riwayat keluarga dan adanya penyakit dasar.
Distrofi korneamakula: Pewarisan resesif autosomal akibat mutasi gen CHST6, disertai kekeruhan difus seperti kaca buram dan kelainan endotel.
Distrofi kornea tetesan koloid: Pewarisan resesif autosomal akibat mutasi gen TACSTD2, ditandai dengan tonjolan koloid seperti susu. Cukup sering ditemukan di Jepang.
QMengapa tes genetik penting?
A
Pada distrofi kornea kisi, meskipun fenotipnya mirip, perbedaan gen penyebab dan lokasi mutasi menyebabkan perbedaan signifikan dalam kecepatan progresi, frekuensi kekambuhan, pilihan pengobatan, dan adanya komplikasi sistemik. LCD1 akibat mutasi TGFBI dan sindrom Meretoja akibat mutasi GSN memiliki perbedaan mendasar dalam strategi pengobatan dan kebutuhan pemeriksaan sistemik2,10). Selain itu, telah dilaporkan kasus dengan mutasi de novo yang tidak dapat ditentukan jenis penyakitnya hanya berdasarkan riwayat keluarga1), sehingga tes genetik sangat penting untuk diagnosis pasti dan klasifikasi tipe.
Pada masa kanak-kanak hingga dewasa muda, saat tidak bergejala atau hanya kekeruhan halus, dilakukan observasi. Evaluasi perkembangan dilakukan dengan pemeriksaan slit-lamp setiap 6 bulan hingga 1 tahun.
Untuk erosi kornea berulang, gejala inti LCD1, terapi konservatif berikut adalah langkah pertama:
Terapi serangan: Penggunaan lensa kontak lunak terapeutik secara terus-menerus untuk melindungi epitel kornea. Dikombinasikan dengan tetes mata antibakteri untuk mencegah infeksi sekunder. Salep mata dioleskan untuk lubrikasi dan perlindungan epitel.
Pencegahan kekambuhan: Pemberian salep mata sebelum tidur efektif menekan kekambuhan serangan erosi kornea berulang. Di lingkungan kering, gunakan air mata buatan atau pelumas pada siang hari.
Pada LCD1, di mana deposit amiloid dominan di lapisan superfisial kornea, dan pada kasus kekeruhan sentral berat atau erosi kornea berulang yang sering, keratektomi fototerapeutik (PTK) dengan laser eksimer adalah pilihan pertama7,8). Biasanya tidak terjadi kekambuhan dini, namun kekambuhan seiring waktu tidak dapat dihindari, dan PTK dapat dilakukan pada mata yang sama hingga dua kali.
Pada heterozigot, kekambuhan berlangsung lambat dan jarang memerlukan perawatan ulang. Pada homozigot, cenderung kambuh lebih awal dibandingkan heterozigot. Tingkat kekambuhan setelah PTK meningkat seiring waktu, sama seperti distrofi terkait TGFBI lainnya, dan pada pengamatan jangka panjang, sebagian besar kasus menunjukkan beberapa tanda kekambuhan8).
Sebagai contoh efektivitas PTK, pada kasus LCD IIIA akibat mutasi de novo TGFBI L509P, dilakukan PTK sedalam 60 µm dengan panduan FD-OCT, dan tajam penglihatan terkoreksi terbaik (BCVA) membaik dari 20/400 menjadi 20/501). Pada 45 bulan pasca operasi, tidak ditemukan penurunan tajam penglihatan atau kekambuhan signifikan1).
Menurut AAO Preferred Practice Pattern untuk edema dan kekeruhan kornea, PTK untuk distrofi kornea granular dan lattice merupakan “pilihan yang wajar” dan dapat menunda transisi ke DALK atau transplantasi kornea penetrasi, namun memiliki risiko kekeruhan pasca operasi. Pada pengulangan, penggunaan mitomisin C bersamaan dipertimbangkan sebagai cara untuk menekan jaringan parut berulang atau deposit stroma, dan diperingatkan bahwa risiko ektasia kornea meningkat jika ablasi melebihi sepertiga anterior stroma atau jika sisa dasar kurang dari 250 µm7).
Pada kasus dengan kekambuhan berulang atau kekeruhan yang meluas ke lapisan stroma yang lebih dalam, transplantasi kornea dipilih. Pada LCD1, biasanya transplantasi kornea tidak diperlukan hingga usia di atas 40 tahun. Karena sel endotel kornea pada LCD umumnya normal, teknik bedah dipilih berdasarkan kedalaman kekeruhan.
Pemulihan penglihatan tinggi tetapi risiko penolakan dan kekambuhan
Dalam beberapa tahun terakhir, DALK telah banyak digunakan sebagai pilihan pertama yang baru karena mengurangi risiko penolakan dan memberikan hasil penglihatan yang setara dengan keratoplasti penetrasi.
Kekambuhan LCD setelah transplantasi kornea tidak dapat dihindari, dan tingkat kekambuhan setelah keratoplasti penetrasi dilaporkan sebesar 17,8% dalam 5 tahun, 26% dalam 8 tahun, dan 56% dalam 15 tahun9). Kekeruhan yang kambuh biasanya terbatas pada lapisan superfisial, sehingga dapat dihilangkan dengan PTK dan memperpanjang waktu hingga transplantasi ulang. Untuk LCD IIIA (tipe varian), seringkali tidak memerlukan pengobatan kecuali jika dampaknya terhadap penglihatan sudah parah.
QSeberapa efektif PTK?
A
PTK dapat secara efektif menghilangkan deposisi amiloid superfisial, meningkatkan penglihatan dan mengurangi erosi epitel berulang. Pada kasus LCD IIIA, dilaporkan perbaikan ketajaman penglihatan terkoreksi dari 20/400 menjadi 20/50 setelah PTK 60 µm, tanpa kekambuhan selama 45 bulan1). Pada heterozigot, kekambuhan lambat, tetapi pada homozigot terjadi kekambuhan dini. Lesi dalam tidak dapat dihilangkan dengan PTK, sehingga kekeruhan dalam memerlukan DALK atau keratoplasti penetrasi7).
QApakah penyakit kambuh setelah transplantasi kornea?
A
Kekambuhan LCD setelah transplantasi kornea tidak dapat dihindari. Tingkat kekambuhan setelah keratoplasti penetrasi dilaporkan sebesar 17,8% dalam 5 tahun, 26% dalam 8 tahun, dan 56% dalam 15 tahun9). Namun, kekeruhan yang kambuh biasanya terbatas pada lapisan superfisial cangkok, sehingga dapat dihilangkan dengan PTK dan memperpanjang umur cangkok. Keratoplasti lamellar dalam (DALK) memiliki risiko penolakan endotel yang lebih rendah dibandingkan keratoplasti penetrasi, dan menjadi pilihan pertama yang baru7).
6. Patofisiologi dan Mekanisme Penyakit yang Mendetail
Inti dari patologi LCD1 adalah akumulasi abnormal TGFBIp (kerato-epithelin, βig-h3). TGFBIp normalnya diproduksi oleh epitel kornea dan tersebar di seluruh lapisan kornea, serta berperan sebagai protein struktural yang terlibat dalam pembangunan serat kolagen dan adhesi sel di stroma 5). Protein abnormal yang dihasilkan oleh mutasi R124C mengalami misfolding dan agregasi diri, mengendap sebagai fibril amiloid yang tidak larut di lapisan Bowman hingga stroma superfisial. Pada tahap lanjut, endapan menyebar ke stroma dalam.
Endapan amiloid menyebabkan perubahan pada struktur adhesi epitel kornea anterior, mengakibatkan degenerasi sel basal epitel dan degenerasi lapisan epitel yang disertai defek membran Bowman. Kerusakan struktural ini menjadi dasar patofisiologi erosi epitel kornea rekuren.
Pada gen TGFBI, perbedaan lokasi mutasi dan asam amino pengganti menentukan gambaran klinis. R124C menyebabkan LCD1, R124H menyebabkan distrofi kornea granular tipe II (tipe Avellino), dan R124L menyebabkan distrofi kornea Reis-Bücklers 5). Mekanisme molekuler bagaimana perbedaan satu asam amino menentukan bahan endapan (amiloid vs hialin vs keduanya) dan lokasi endapan belum sepenuhnya dipahami, namun diyakini bahwa lokasi mutasi dalam domain βig-h3 dan pengaruhnya terhadap stabilitas lipatan adalah kuncinya.
Pada LCD IIIA, mutasi dominan lapisan dalam seperti L527R menghasilkan garis kisi tebal seperti tali, dan menjadi tipe onset lambat tanpa gangguan epitel. Lokalisasi endapan berlapis dapat dijelaskan oleh gradien sekresi dan difusi βig-h3 dari sel penghasil (sel basal epitel) ke stroma, serta perbedaan stabilitas lipatan protein mutan. R124C diyakini memprioritaskan jalur dari intermediate lipatan menuju pembentukan fibril amiloid, mengakumulasi amiloid di sekitar lapisan Bowman5). Sementara itu, mutasi L527R membentuk protein misfold yang relatif stabil dan mengendap perlahan di stroma yang lebih dalam.
Amiloid kornea posterior dan risiko operasi intraokular
Secara tradisional, endapan amiloid pada LCD1 dianggap terbatas pada kornea anterior (lapisan Bowman hingga stroma superfisial). Namun, pemeriksaan patologis terbaru menunjukkan adanya endapan amiloid di kornea posterior dekat membran Descemet3). Endapan amiloid di kornea posterior dapat mempengaruhi adhesi membran Descemet, berkontribusi pada pelepasan membran Descemet selama operasi katarak3). Diduga mekanisme yang sama yang mengganggu adhesi epitel di kornea anterior juga bekerja di bagian posterior 3).
Gelsolin, molekul penyebab sindrom Meretoja (sebelumnya LCD2), terdapat di sitoplasma dan ekstraseluler, dan merupakan protein yang terlibat dalam motilitas sel, pembelahan sel, dan apoptosis melalui pengikatan aktin. Mutasi klasik D187N, yang disebut tipe Finlandia, menyebabkan deposit kisi kornea dan neuropati saraf kranial sebagai fenotipe utama11). Mutasi baru p.Glu580Lys yang dilaporkan pada keluarga Slovenia terletak di batas domain G4-G5, dan substitusi asam glutamat bermuatan negatif menjadi lisin bermuatan positif menyebabkan tolakan elektrostatik, mengurangi konektivitas dan stabilitas antar domain2). Gelsolin mutan mengalami pemotongan abnormal oleh furin dan MT1-MMP dalam plasma, melepaskan fragmen prekursor amiloid 8 kDa dan 5 kDa. Fragmen-fragmen ini mengendap di stroma kornea, kulit, dinding pembuluh darah, saraf perifer, dan glomerulus ginjal, menyebabkan gejala multi-organ yang khas dari sindrom Meretoja2,11). Deposit kornea sering mendahului gejala sistemik lainnya, sehingga dokter mata dapat menjadi yang pertama mendiagnosis penyakit ini.
Telah dilaporkan terjadinya LCD akibat mutasi de novo pada gen TGFBI1). Bahkan pada kasus tanpa riwayat keluarga, kemungkinan mutasi de novo harus dipertimbangkan, dan konfirmasi melalui tes genetik dianjurkan1). Mutasi L509P jarang terjadi tetapi menyebabkan fenotipe yang bervariasi dari distrofi kornea Reis-Bücklers hingga mirip LCD IIIA1).
Pada gen GSN, selain mutasi tradisional p.Asp187Asn/Tyr, telah dilaporkan mutasi baru p.Glu580Lys yang menyebabkan amiloidosis sistemik dengan distrofi kisi kornea, kulit kendur, aritmia jantung, gangguan ginjal, dan neuropati optik2).
Lesi kornea posterior dan manajemen bedah intraokular
Studi patologis menunjukkan adanya deposit amiloid di kornea posterior pasien LCD1, yang dapat mempengaruhi adhesi membran Descemet3). Perhatian diperlukan terhadap risiko pelepasan membran Descemet selama operasi intraokular, seperti operasi katarak.
Temuan ini memiliki implikasi klinis dalam evaluasi indikasi operasi katarak dan perencanaan teknik bedah pada pasien LCD1.
Prosedur bedah yang lebih presisi seperti keratektomi lamelar berbantuan laser femtosecond (femtosecond laser-assisted lamellar keratectomy, FLK) dan keratoplasti lamelar berbantuan laser femtosecond (femtosecond laser-assisted lamellar keratoplasty, FALK) sedang dikembangkan 12). Prosedur-prosedur ini diposisikan sebagai pilihan pelengkap untuk PTK konvensional karena meningkatkan kehalusan permukaan eksisi dan kontrol kedalaman yang sangat reprodusibel.
Karena mutasi TGFBI merupakan mutasi dominan autosomal dengan gain-of-function, siRNA spesifik alel mutan, oligonukleotida antisense, dan knockout spesifik alel dengan CRISPR-Cas9 sedang diteliti pada tahap studi praklinis. Kornea merupakan organ target yang menguntungkan untuk terapi gen karena dapat diberikan secara topikal dan memiliki hak istimewa imun. Namun, saat ini belum ada yang diaplikasikan secara klinis, dan semuanya memerlukan verifikasi keamanan dan efektivitas jangka panjang di masa depan.
Penghambat Pembentukan Amiloid dan Chaperon Molekuler
Senyawa molekul kecil yang menargetkan proses agregasi TGFBIp dan gelsolin mutan, chaperon molekuler (seperti penginduksi Hsp70), dan penghambat pengikatan fibril amiloid sedang diteliti pada tahap penelitian dasar. Untuk amiloidosis tipe gelsolin sistemik, obat yang menghambat tahap pemotongan gelsolin mutan dalam plasma telah dievaluasi dalam beberapa uji praklinis 2). Di masa depan, terapi target molekuler semacam ini diharapkan dapat menjadi terapi kuratif yang menggantikan eksisi fisik konvensional (PTK dan transplantasi kornea).
Analisis proteom kornea menggunakan spektrometri massa menunjukkan bahwa deposit LCD1 mungkin mengandung kopresipitasi beberapa protein abnormal selain TGFBIp. Kontribusi patologis dari protein kopresipitasi ini sedang diteliti untuk aplikasi klinis di masa depan.
Ji YW, Ahn H, Shin KJ, Kim TI, Seo KY, Stulting RD, et al. De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. Journal of clinical medicine. 2022;11(11). doi:10.3390/jcm11113055. PMID:35683443; PMCID:PMC9181583.
Potrc M, Volk M, de Rosa M, Pizem J, Teran N, Jaklic H, Maver A, Drnovsek-Olup B, Bollati M, Vogelnik K, Hocevar A, Gornik A, Pfeifer V, Peterlin B, Hawlina M, Fakin A. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. Int J Mol Sci. 2021;22(3):1084.
Matsumoto A, Fukuoka H, Sotozono C. Descemet’s Membrane Detachment During Cataract Surgery in Lattice Corneal Dystrophy Type I: Histopathological Analysis of Posterior Corneal Involvement. Cureus. 2025;17(3):e81431. doi:10.7759/cureus.81431. PMID:40296953; PMCID:PMC12037203.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
Lisch W, Seitz B. The clinical landmarks of corneal dystrophies. Developments in ophthalmology. 2011;48:9-23. doi:10.1159/000324075. PMID:21540629.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern. San Francisco, CA: American Academy of Ophthalmology; 2024.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea. 2003;22(1):19-21.
Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969;1(4):314-324. PMID:4313418.
Kivelä T, Tarkkanen A, Frangione B, Ghiso J, Haltia M. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja’s syndrome. Invest Ophthalmol Vis Sci. 1994;35(10):3759-3769. PMID:8088963.
Steger B, Romano V, Biddolph S, Willoughby CE, Batterbury M, Kaye SB. Femtosecond laser-assisted lamellar keratectomy for corneal opacities secondary to anterior corneal dystrophies: an interventional case series. Cornea. 2016;35(1):6-13. PMID:26509759. doi:10.1097/ICO.0000000000000665.
Salin teks artikel dan tempelkan ke asisten AI pilihan Anda.
Artikel disalin ke papan klip
Buka asisten AI di bawah, lalu tempelkan teks yang disalin ke kotak chat.