La distrofia corneale a reticolo (lattice corneal dystrophy, LCD) è una distrofia corneale ereditaria in cui l’amiloide si deposita nello stroma corneale, causando opacità lineari a forma di reticolo. Descritta per la prima volta negli anni 1890, è una malattia di antica conoscenza. Nella classificazione clinico-genetica della seconda edizione IC3D (International Committee for Classification of Corneal Dystrophies) è stata unificata in LCD1 e le sue varianti (tipi 3, 3A, 1/3A e 4 della classificazione tradizionale)4).
LCD1, la distrofia corneale granulare, la distrofia corneale di Reis-Bücklers e la distrofia corneale di Thiel-Behnke formano un gruppo di malattie note come «distrofie correlate a TGFBI». Il gene responsabile TGFBI (transforming growth factor beta-induced gene) si trova sul braccio lungo del cromosoma 5 (5q31) e la trasmissione è autosomica dominante. La proteina TGFBI (TGFBIp, cheratoepitelina, βig-h3) è prodotta dall’epitelio corneale e distribuita in tutto lo spessore della cornea. Nello stroma corneale partecipa all’organizzazione delle fibre di collagene. Mutazioni diverse dello stesso gene, a seconda del sito e della sostituzione amminoacidica, portano a differenze significative nel materiale depositato (ialino o amiloide) e nel quadro clinico5).
La mutazione rappresentativa di LCD1 è R124C, in cui l’arginina in posizione 124 del gene TGFBI viene sostituita da cisteina. Nella variante LCD IIIA sono state riportate mutazioni come L527R.
La proteina anomala accumulata nella cornea si colora di rosso con la colorazione rosso Congo e mostra una birifrangenza verde mela caratteristica al microscopio a luce polarizzata, confermando così la presenza di amiloide. Questo reperto è un indicatore diagnostico tissutale classico dell’amiloidosi sin dal XIX secolo6).
Il tipo precedentemente chiamato “distrofia corneale reticolare di tipo 2” è una manifestazione oculare dell’amiloidosi sistemica da gelsolina (GSN-AMYL, sindrome di Meretoja). Nella classificazione IC3D attuale è classificato come “amiloidosi familiare” e viene trattato separatamente dalla LCD classica4,10). Descritta per la prima volta da Meretoja in Finlandia nel 1969, questa sindrome è una malattia ereditaria caratterizzata da opacità corneali reticolari, neuropatia cranica progressiva, lassità cutanea e sintomi sistemici10,11). Poiché la diagnosi differenziale tra le due condizioni è importante nella pratica clinica, questo articolo le descrive entrambe.
In Giappone, la distrofia correlata a TGFBI più frequente è di gran lunga il tipo granulare II (Avellino, R124H), mentre la LCD1 è meno comune. Tuttavia, poiché entrambe derivano da differenze di pochi nucleotidi nello stesso gene TGFBI, in caso di sovrapposizione clinica è consigliabile la conferma tramite test genetico. La prevalenza esatta della LCD in Giappone non è stata riportata, ma rientra in una categoria relativamente rara tra le distrofie corneali.
QQual è la differenza tra LCD1 e sindrome di Meretoja?
A
La LCD1 è un deposito di amiloide limitato alla cornea causato da mutazioni del gene TGFBI, che insorge nella zona pupillare tra i 10 e i 20 anni ed è frequentemente associata a erosioni epiteliali ricorrenti. La sindrome di Meretoja (ex LCD2, tipo GSN), invece, è una manifestazione oculare di amiloidosi sistemica dovuta a mutazioni del gene GSN (gelsolina), che insorge nella cornea periferica tra i 30 e i 40 anni, mantenendo a lungo la trasparenza centrale. La sindrome di Meretoja si accompagna a sintomi sistemici come lassità cutanea, facies maschera, neuropatia periferica e aritmie cardiache2,10). Nella seconda edizione IC3D, la sindrome di Meretoja è classificata indipendentemente dalla distrofia corneale reticolare4).
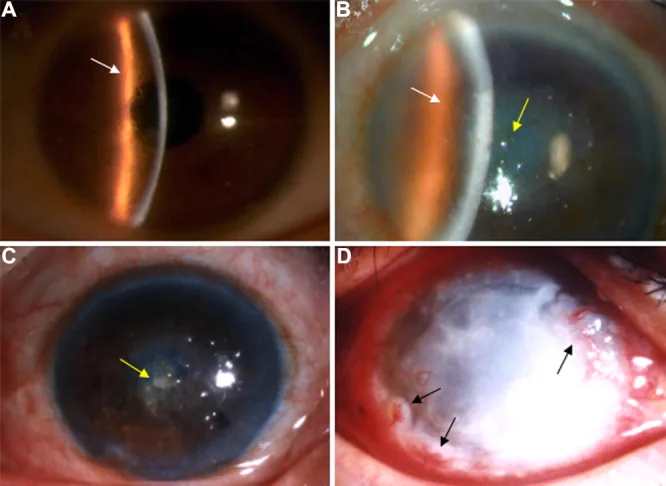
Liu Z, et al. An R124C mutation in TGFBI caused lattice corneal dystrophy type I with a variable phenotype in three Chinese families. Mol Vis. 2008. Figure 2. PMCID: PMC2443752. License: CC BY.
Nella fotografia con lampada a fessura, si osservano linee reticolari ramificate nello stroma corneale e opacità predominanti nella parte centrale. L’immagine mostra i reperti clinici tipici della distrofia corneale reticolare.
Nella LCD1, molti pazienti sono asintomatici durante l’infanzia, con solo lievi opacità rilevabili con l’illuminazione a fessura della lampada a fessura. Dopo i 10-20 anni, si verificano episodi ricorrenti di erosione corneale (RCE), con dolore oculare acuto al risveglio, fotofobia, lacrimazione e sensazione di corpo estraneo. Intorno ai 30 anni, compaiono opacità bianche nello stroma superficiale centrale della cornea, e dopo i 40 anni la visione diminuisce progressivamente.
Nella LCD IIIA (variante), di solito non si verificano danni epiteliali e il sintomo principale è una lenta diminuzione della vista dopo i 40 anni.
Nella vecchia LCD2 (sindrome di Meretoja), i sintomi oculari compaiono tra i 30 e i 40 anni, ma un significativo deficit visivo spesso si manifesta solo dopo i 60 anni11). Spesso precedono o accompagnano sintomi sistemici come lassità cutanea palpebrale, facies a maschera, neuropatia cranica progressiva e aritmia cardiaca2,10).
Di seguito sono riportati i reperti alla lampada a fessura per ciascun tipo.
LCD1 (classica)
Sede iniziale: Appare come sottili opacità puntiformi e lineari nello strato di Bowman e nello stroma superficiale nell’area pupillare di entrambi gli occhi.
Linee reticolari: Opacità filamentose e lineari con doppio contorno si intrecciano formando opacità reticolari e stellari.
Fase avanzata: Si sviluppa un’opacità bianco-lattea di forma ovale o rotonda nella cornea centrale.
Illuminazione a riflessione: Le sottili linee reticolari semitrasparenti, difficili da vedere con illuminazione diretta, diventano chiaramente visibili.
Colorazione con fluoresceina: La ridotta adesione epiteliale rende la superficie ruvida.
Erosione epiteliale ricorrente: Si verifica frequentemente perché i depositi coinvolgono le cellule basali dell’epitelio e la membrana di Bowman.
LCD IIIA (variante)
Linee reticolari: linee reticolari spesse e lunghe nello stroma medio-profondo, talvolta con ramificazioni dendritiche. Osservabili anche con illuminazione diretta.
Fenotipo: esistono tre pattern: ① solo linee reticolari, ② solo depositi granulari, ③ misti. Possono presentarsi fenotipi diversi tra occhio destro e sinistro nello stesso individuo, o casi unilaterali.
Epitelio: di solito non si verificano alterazioni epiteliali.
Omozigoti: negli omozigoti L527R le linee reticolari sono più spesse e i depositi granulari centrali più grandi, ma la differenza non è così marcata come tra eterozigoti e omozigoti per R124H (tipo granulare II).
Tipo GSN (Meretoja)
Linee reticolari: pochi depositi reticolari, poco fini, che appaiono radialmente dalla periferia.
Trasparenza centrale: la trasparenza centrale si mantiene a lungo dopo l’esordio.
Erosione epiteliale: rara.
Reperti sistemici: si presentano alterazioni del volto come facies a maschera, labbra prominenti con disturbi motori, orecchie cadenti, e blefarocalasia 2).
Nella LCD1, in alcuni casi l’opacità circolare centrale può essere particolarmente marcata; è stato riportato un caso di un eterozigote R124C di 56 anni con opacità circolare centrale che ha richiesto il trapianto di cornea.
QSi può diagnosticare la LCD1 anche nei bambini?
A
La LCD1 in età pediatrica è spesso asintomatica e le anomalie sono difficili da rilevare con la sola illuminazione diretta. Con un esame dettagliato mediante illuminazione a fessura con retroilluminazione o illuminazione indiretta, si possono identificare fini opacità puntiformi o lineari nello stroma superficiale centrale. Nei bambini con erosioni epiteliali corneali ricorrenti, si raccomanda di considerare la LCD1, raccogliere la storia familiare e valutare le cornee dei genitori. Il test genetico per TGFBI è utile per la diagnosi definitiva.
Modalità di trasmissione: Ereditarietà autosomica dominante.
Mutazione tipica LCD1: R124C (Arg124Cys) è la più frequente5).
Mutazione tipica LCD IIIA: L527R (Leu527Arg) e altre sono state riportate. Esistono anche casi omozigoti.
Mutazione de novo: È stato riportato un caso di mutazione de novo TGFBI L509P che ha presentato il fenotipo LCD IIIA1). I genitori non presentavano la mutazione, mentre è stata ereditata da uno dei figli1).
Ruolo di TGFBIp: Prodotto dall’epitelio corneale, si distribuisce in tutto lo spessore della cornea; nello stroma partecipa all’organizzazione delle fibre di collagene5).
Correlato a GSN (sindrome di Meretoja, ex LCD2)
Locus genico: 9q34 (gene GSN, gelsolina).
Modalità di trasmissione: Ereditarietà autosomica dominante.
Mutazione classica: D187N (tipo finlandese) è la più comune; è stata riportata anche p.Asp187Tyr10,11).
Nuova mutazione: p.Glu580Lys, riportata in una famiglia slovena, si trova al confine tra i domini G4-G5 e provoca repulsione elettrostatica a causa della sostituzione di una carica negativa con una positiva2).
Quadro clinico: Oltre all’opacità corneale a reticolo, si manifesta con amiloidosi sistemica che comprende lassità cutanea, aritmie cardiache, danno renale e neuropatia ottica2).
Poiché si tratta di una malattia ereditaria, la storia familiare è il fattore di rischio più importante. Tuttavia, per TGFBI possono verificarsi mutazioni de novo, quindi l’assenza di storia familiare non è sufficiente per escluderla1). La modalità di trasmissione è autosomica dominante: se uno dei genitori è portatore della mutazione, il figlio ha una probabilità del 50% di ereditarla. Non si osservano differenze di genere, e per LCD1 non è evidente una differenza razziale, mentre la sindrome di Meretoja è nota per l’accumulo di famiglie numerose in Finlandia11).
Il contributo dei fattori ambientali non è chiaro; l’insorgenza e la progressione di questa malattia sono determinate principalmente dal genotipo. Tuttavia, la frequenza delle erosioni epiteliali ricorrenti può aumentare in ambienti secchi, con l’uso di lenti a contatto o in seguito a traumi. La chirurgia refrattiva (LASIK, SMILE, ecc.) può causare un rapido peggioramento delle distrofie corneali correlate a TGFBI; pertanto, nei casi con storia familiare, è necessaria attenzione nello screening preoperatorio5).
Per la diagnosi differenziale tra LCD1, le varianti e il tipo GSN, si devono integrare i reperti della lampada a fessura, i reperti istologici e i reperti genetici.
Esami clinici
Microscopio a lampada a fessura: Con illuminazione diretta, le linee reticolari iniziali sono facili da trascurare. Con la tecnica di transilluminazione, si rilevano le opacità fini sullo sfondo della pupilla; con l’illuminazione a retroilluminazione, si rilevano le sottili linee reticolari semitrasparenti.
Colorazione con fluoresceina: Nella LCD1, a causa della ridotta adesione epiteliale, la colorazione appare ruvida. È utile anche per valutare l’estensione dell’erosione epiteliale.
Tomografia a coerenza ottica del segmento anteriore (OCT anteriore): Consente di valutare quantitativamente la profondità stratificata dei depositi. La misurazione della profondità della lesione con FD-OCT è utile per determinare la profondità di escissione nella PTK1).
Microscopia confocale corneale: Consente di osservare i depositi nello stroma a livello cellulare.
Diagnosi definitiva
Test genetico: La rilevazione di mutazioni nei geni TGFBI e GSN conferma il tipo di malattia. Poiché mutazioni diverse nello stesso fenotipo possono alterare la velocità di recidiva e progressione, ciò è direttamente collegato alla pianificazione del trattamento.
Esame patologico: La colorazione con rosso Congo mostra una colorazione rossa e, al microscopio a luce polarizzata, mostra birifrangenza verde mela, confermando l’amiloide6).
Immunoistochimica: È possibile la differenziazione del tipo di malattia mediante anticorpi anti-TGFBIp e anti-gelsolina.
Anamnesi familiare: Poiché la trasmissione è autosomica dominante, la conferma dei reperti corneali nei genitori e nei fratelli supporta la diagnosi.
Distrofia corneale granulare di tipo II (tipo Avellino, TGFBI R124H): È la distrofia correlata a TGFBI più comune in Giappone e mostra un quadro misto di depositi granulari e linee reticolari. Per la diagnosi differenziale con LCD1, il test genetico è affidabile.
Amiloidosi corneale secondaria: non ereditaria, l’amiloide si deposita secondariamente a stimoli cronici della superficie oculare come trichiasi o cheratocono. L’assenza di storia familiare e la presenza di una malattia di base sono elementi differenziali.
Distrofia corneale maculare: eredità autosomica recessiva da mutazione del gene CHST6, con opacità diffusa a vetro smerigliato e anomalie endoteliali.
Distrofia corneale gelatinosa a goccia: eredità autosomica recessiva da mutazione del gene TACSTD2, con rilievi gelatinosi bianco latte. Relativamente comune in Giappone.
QPerché il test genetico è importante?
A
Nella distrofia corneale a reticolo, anche se il fenotipo è simile, geni causali e siti di mutazione diversi comportano grandi differenze nella velocità di progressione, frequenza di recidiva, scelta terapeutica e presenza di complicanze sistemiche. La LCD1 da mutazione TGFBI e la sindrome di Meretoja da mutazione GSN differiscono radicalmente nella strategia terapeutica e nella necessità di indagini sistemiche2,10). Inoltre, sono stati riportati casi con mutazioni de novo in cui la sola storia familiare non è sufficiente a determinare il tipo di malattia1), rendendo il test genetico indispensabile per la diagnosi definitiva e la classificazione.
Nell’infanzia e nella giovinezza, in fase asintomatica o con solo lievi opacità, si procede con osservazione. Valutare la progressione con esame alla lampada a fessura ogni 6-12 mesi.
Per l’erosione epiteliale ricorrente, sintomo centrale della LCD1, la prima linea è la terapia conservativa.
Trattamento dell’attacco acuto: uso continuo di lenti a contatto morbide terapeutiche per proteggere l’epitelio corneale. Associare colliri antibiotici per prevenire infezioni secondarie. Applicare unguenti oculari per lubrificazione e protezione epiteliale.
Prevenzione delle recidive: la somministrazione di unguento oculare prima di dormire è efficace nel ridurre le recidive di RCE. In ambienti secchi, utilizzare lacrime artificiali o lubrificanti anche durante il giorno.
Nella LCD1, caratterizzata da depositi di amiloide nello strato corneale superficiale, in caso di opacità centrale marcata o erosioni epiteliali ricorrenti, la cheratectomia fototerapeutica (PTK) con laser ad eccimeri è la prima scelta7,8). Di solito non si verificano recidive precoci, ma la recidiva è inevitabile nel tempo; il trattamento PTK può essere eseguito fino a circa due volte sullo stesso occhio.
Negli eterozigoti la recidiva è lenta e raramente richiede un nuovo trattamento. Negli omozigoti la recidiva tende a manifestarsi più precocemente rispetto agli eterozigoti. Il tasso di recidiva dopo PTK aumenta nel tempo, come per altre distrofie corneali correlate a TGFBI, e in molti casi si osservano segni di recidiva durante il follow-up a lungo termine8).
Un caso che dimostra l’efficacia della PTK è quello di un paziente con LCD IIIA dovuta a una mutazione de novo TGFBI L509P, in cui è stata eseguita una PTK di 60 µm guidata da FD-OCT, con miglioramento della migliore acuità visiva corretta (BCVA) da 20/400 a 20/501). A 45 mesi dall’intervento non sono state osservate riduzione della vista o recidive significative1).
Secondo il Preferred Practice Pattern dell’AAO sull’edema e l’opacità corneale, la PTK per la distrofia corneale granulare e reticolare è una ‘opzione ragionevole’ e può ritardare il passaggio a DALK o cheratoplastica perforante, ma comporta il rischio di haze post-operatorio. In caso di ripetizione, l’uso concomitante di mitomicina C è considerato un mezzo per sopprimere la cicatrizzazione ricorrente e i depositi stromali. Si avverte che il rischio di ectasia corneale aumenta se l’ablazione supera il terzo anteriore dello stroma o se il letto residuo è inferiore a 250 µm7).
Nei casi di recidiva frequente o quando l’opacità si estende oltre lo stroma medio-profondo, si opta per la cheratoplastica. Nella LCD1, di solito non si ricorre alla cheratoplastica prima dei 40 anni. Poiché le cellule endoteliali corneali sono generalmente normali nella LCD, la tecnica chirurgica viene scelta in base alla profondità dell’opacità.
Elevato recupero visivo ma rischio di rigetto e recidiva
Negli ultimi anni, la DALK è diventata ampiamente utilizzata come nuova prima scelta grazie alla riduzione del rischio di rigetto e ai risultati visivi paragonabili alla cheratoplastica penetrante.
La recidiva di LCD dopo cheratoplastica è un fenomeno inevitabile; il tasso di recidiva dopo cheratoplastica penetrante è riportato essere del 17,8% a 5 anni, del 26% a 8 anni e del 56% a 15 anni9). Poiché l’opacità recidivante è solitamente limitata agli strati superficiali, può essere rimossa con PTK, prolungando l’intervallo prima di un nuovo trapianto. Per LCD IIIA (variante), spesso non è necessario alcun trattamento a meno che non vi sia un impatto significativo sulla vista.
QQuanto è efficace la PTK?
A
La PTK può rimuovere efficacemente i depositi di amiloide superficiali, migliorando la vista e riducendo le erosioni epiteliali ricorrenti. In un caso di LCD IIIA, dopo PTK di 60 µm, la migliore acuità visiva corretta è migliorata da 20/400 a 20/50 e non si è verificata recidiva per 45 mesi1). Negli eterozigoti la recidiva è lenta, mentre negli omozigoti si osserva una recidiva precoce. Le lesioni profonde non possono essere rimosse con PTK, quindi per le opacità profonde è necessaria DALK o cheratoplastica penetrante7).
QLa LCD recidiva dopo cheratoplastica?
A
La recidiva di LCD dopo cheratoplastica è inevitabile. Il tasso di recidiva dopo cheratoplastica penetrante è riportato essere del 17,8% a 5 anni, del 26% a 8 anni e del 56% a 15 anni9). Tuttavia, poiché l’opacità recidivante è solitamente limitata agli strati superficiali del trapianto, può essere rimossa con PTK, prolungando la durata del trapianto. La cheratoplastica lamellare profonda (DALK) presenta un rischio inferiore di rigetto endoteliale rispetto alla cheratoplastica penetrante ed è considerata una nuova prima scelta7).
Il centro della patologia di LCD1 è l’accumulo anomalo di TGFBIp (cherato-epitelina, βig-h3). TGFBIp è una proteina strutturale normalmente prodotta dall’epitelio corneale, distribuita in tutto lo spessore della cornea, e coinvolta nella costruzione delle fibre di collagene e nell’adesione cellulare nello stroma 5). La proteina anomala prodotta dalla mutazione R124C subisce un misfolding e un’auto-aggregazione, depositandosi come fibrille amiloidi insolubili nello strato di Bowman e nello stroma superficiale. Nelle fasi avanzate, il deposito si estende in profondità nello stroma.
Il deposito amiloide altera le strutture di adesione epiteliale nella cornea anteriore, causando degenerazione delle cellule basali epiteliali e degenerazione dello strato epiteliale con perdita della membrana di Bowman. Questa rottura strutturale costituisce la base patofisiologica delle erosioni epiteliali corneali ricorrenti.
Differenziazione fenotipica in base al genotipo TGFBI
Nel gene TGFBI, la sede della mutazione e la sostituzione aminoacidica determinano il quadro clinico. R124C causa LCD1, R124H causa la distrofia corneale granulare di tipo II (tipo Avellino), e R124L causa la distrofia corneale di Reis-Bücklers 5). Il meccanismo molecolare per cui una singola differenza aminoacidica determina la sostanza depositata (amiloide vs ialina vs entrambe) e la sede del deposito non è completamente chiarito, ma si ritiene che la sede della mutazione nel dominio di βig-h3 e l’effetto sulla stabilità del ripiegamento siano fattori chiave.
Nella LCD IIIA, mutazioni profonde come L527R producono linee lattice spesse a forma di corda, con una forma tardiva senza disturbi epiteliali. La localizzazione stratificata dei depositi può essere spiegata dal gradiente di secrezione e diffusione di βig-h3 dalle cellule produttrici (cellule basali epiteliali) nello stroma e dalle differenze nella stabilità di ripiegamento della proteina mutata. Si ritiene che R124C favorisca il percorso dall’intermedio di ripiegamento alla formazione di fibrille amiloidi, accumulando amiloide intorno allo strato di Bowman 5). D’altra parte, la mutazione L527R forma una proteina misfolded relativamente stabile, che si deposita lentamente negli strati stromali più profondi.
Amiloide corneale posteriore e rischio chirurgico intraoculare
Tradizionalmente si riteneva che il deposito amiloide nella LCD1 fosse limitato alla cornea anteriore (strato di Bowman e stroma superficiale). Tuttavia, recenti studi patologici hanno dimostrato la presenza di depositi amiloidi anche nella cornea posteriore vicino alla membrana di Descemet3). Il deposito amiloide nella cornea posteriore potrebbe influenzare l’adesione della membrana di Descemet e contribuire al distacco della stessa durante la chirurgia della cataratta3). Si suggerisce che lo stesso meccanismo con cui il deposito amiloide compromette l’adesione epiteliale nella cornea anteriore agisca anche nella parte posteriore 3).
Meccanismo molecolare dell’amiloidosi di tipo gelsolina
La gelsolina, molecola responsabile della vecchia LCD2 (sindrome di Meretoja), è presente sia nel citoplasma che nello spazio extracellulare ed è una proteina coinvolta nella motilità cellulare, nella divisione cellulare e nell’apoptosi attraverso il legame con l’actina. La classica mutazione D187N, chiamata variante finlandese, causa un fenotipo caratterizzato da depositi corneali a reticolo e neuropatia cranica11). La nuova mutazione p.Glu580Lys, riportata in una famiglia slovena, si trova al confine tra i domini G4-G5 e, sostituendo un acido glutammico carico negativamente con una lisina carica positivamente, genera repulsione elettrostatica, riducendo la connettività e la stabilità tra i domini2). La gelsolina mutante subisce un taglio anomalo nel plasma da parte di furina e MT1-MMP, rilasciando frammenti precursori dell’amiloide di 8 kDa e 5 kDa. Questi si depositano nello stroma corneale, nella pelle, nelle pareti vascolari, nei nervi periferici e nei glomeruli renali, causando i sintomi multiorgano caratteristici della sindrome di Meretoja2,11). I depositi corneali spesso precedono altri sintomi sistemici, e l’oftalmologo può essere il primo a diagnosticare questa malattia.
Sono stati riportati casi di LCD causati da mutazioni de novo del gene TGFBI1). Anche in assenza di storia familiare, è opportuno considerare la possibilità di una mutazione de novo e si raccomanda la conferma tramite test genetici1). La mutazione L509P è rara, ma può presentare fenotipi variabili, dalla distrofia corneale di Reis-Bücklers a quella simile alla LCD IIIA1).
Nel gene GSN, oltre alla classica mutazione p.Asp187Asn/Tyr, è stata riportata una nuova mutazione p.Glu580Lys, che causa amiloidosi sistemica con distrofia corneale a reticolo, lassità cutanea, aritmie cardiache, nefropatia e neuropatia ottica2).
Patologia corneale posteriore e gestione chirurgica intraoculare
È stato dimostrato patologicamente che nei pazienti con LCD1 sono presenti depositi di amiloide nella cornea posteriore, che possono influenzare l’adesione della membrana di Descemet3). È necessario prestare attenzione al rischio di distacco della membrana di Descemet durante la chirurgia intraoculare, inclusa la chirurgia della cataratta.
Questa scoperta ha implicazioni cliniche per la valutazione dell’idoneità alla chirurgia della cataratta e la pianificazione dell’intervento nei pazienti con LCD1.
Si stanno sviluppando tecniche chirurgiche più precise come la cheratectomia lamellare assistita da laser a femtosecondi (femtosecond laser-assisted lamellar keratectomy, FLK) e la cheratoplastica lamellare assistita da laser a femtosecondi (femtosecond laser-assisted lamellar keratoplasty, FALK)12). Queste si stanno affermando come opzioni complementari alla PTK tradizionale, grazie al miglioramento della levigatezza della superficie di resezione e al controllo della profondità altamente riproducibile.
Poiché la mutazione TGFBI è una mutazione autosomica dominante con gain-of-function, siRNA specifici per l’allele mutato, oligonucleotidi antisenso e knockout allele-specifico tramite CRISPR-Cas9 sono in fase di studio preclinico. La cornea è un organo bersaglio favorevole per la terapia genica grazie alla possibilità di somministrazione locale e al privilegio immunitario. Tuttavia, al momento non esistono applicazioni cliniche e tutte richiedono future verifiche di sicurezza ed efficacia a lungo termine.
Farmaci inibitori della formazione di amiloide e chaperoni molecolari
Composti a basso peso molecolare che mirano al processo di aggregazione di TGFBIp e gelsolina mutata, chaperoni molecolari (come induttori di Hsp70) e inibitori del legame delle fibrille amiloidi sono in fase di ricerca di base. Per l’amiloidosi sistemica di tipo gelsolina, alcuni farmaci che inibiscono la fase di scissione della gelsolina mutata nel plasma sono stati valutati in studi preclinici2). In futuro, si prevede che tali terapie molecolari mirate possano diventare un trattamento radicale in sostituzione della tradizionale asportazione fisica (PTK e trapianto di cornea).
L’analisi del proteoma corneale mediante spettrometria di massa ha mostrato che nei depositi di LCD1 non solo TGFBIp ma anche multiple proteine anomale possono co-precipitare. Per future applicazioni cliniche, si sta studiando il contributo patogenetico di queste proteine co-precipitate.
Ji YW, Ahn H, Shin KJ, Kim TI, Seo KY, Stulting RD, et al. De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. Journal of clinical medicine. 2022;11(11). doi:10.3390/jcm11113055. PMID:35683443; PMCID:PMC9181583.
Potrc M, Volk M, de Rosa M, Pizem J, Teran N, Jaklic H, Maver A, Drnovsek-Olup B, Bollati M, Vogelnik K, Hocevar A, Gornik A, Pfeifer V, Peterlin B, Hawlina M, Fakin A. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. Int J Mol Sci. 2021;22(3):1084.
Matsumoto A, Fukuoka H, Sotozono C. Descemet’s Membrane Detachment During Cataract Surgery in Lattice Corneal Dystrophy Type I: Histopathological Analysis of Posterior Corneal Involvement. Cureus. 2025;17(3):e81431. doi:10.7759/cureus.81431. PMID:40296953; PMCID:PMC12037203.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
Lisch W, Seitz B. The clinical landmarks of corneal dystrophies. Developments in ophthalmology. 2011;48:9-23. doi:10.1159/000324075. PMID:21540629.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern. San Francisco, CA: American Academy of Ophthalmology; 2024.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea. 2003;22(1):19-21.
Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969;1(4):314-324. PMID:4313418.
Kivelä T, Tarkkanen A, Frangione B, Ghiso J, Haltia M. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja’s syndrome. Invest Ophthalmol Vis Sci. 1994;35(10):3759-3769. PMID:8088963.
Steger B, Romano V, Biddolph S, Willoughby CE, Batterbury M, Kaye SB. Femtosecond laser-assisted lamellar keratectomy for corneal opacities secondary to anterior corneal dystrophies: an interventional case series. Cornea. 2016;35(1):6-13. PMID:26509759. doi:10.1097/ICO.0000000000000665.
Copia il testo dell'articolo e incollalo nell'assistente IA che preferisci.
Articolo copiato negli appunti
Apri un assistente IA qui sotto e incolla il testo copiato nella chat.