La distrofia corneal reticular (lattice corneal dystrophy, LCD) es una distrofia corneal hereditaria en la que se deposita amiloide en el estroma corneal, produciendo opacidades lineales en forma de red. Es una enfermedad históricamente antigua descrita en la década de 1890, y en la clasificación clínico-genética de la segunda edición de la IC3D (Comité Internacional para la Clasificación de Distrofias Corneales) se unifica en LCD1 y sus variantes (tipos 3, 3A, 1/3A y 4 anteriores)4).
Posicionamiento como distrofia relacionada con TGFBI
LCD1, la distrofia corneal granular, la distrofia corneal de Reis-Bücklers y la distrofia corneal de Thiel-Behnke forman un grupo de enfermedades como “distrofias relacionadas con TGFBI”. El gen causante TGFBI (gen inducido por el factor de crecimiento transformante beta) se localiza en el brazo largo del cromosoma 5 (5q31) y sigue un patrón de herencia autosómica dominante. La proteína TGFBI (TGFBIp, queratoepitelina, βig-h3) es producida por las células epiteliales corneales y se distribuye en todo el espesor de la córnea. En el estroma corneal, participa en el ensamblaje de las fibras de colágeno. Incluso con mutaciones en el mismo gen, las diferencias en el sitio de mutación y la sustitución de aminoácidos conducen a una divergencia significativa en el material depositado (hialino o amiloide) y la presentación clínica5).
La mutación representativa de LCD1 es R124C, donde la arginina en la posición 124 del gen TGFBI se reemplaza por cisteína. En la variante LCD IIIA, se han reportado mutaciones como L527R.
Diagnóstico histológico y características clásicas
La proteína anormal acumulada en la córnea se tiñe de rojo con rojo Congo y muestra una birrefringencia verde manzana característica bajo microscopio de luz polarizada, confirmándose como amiloide. Este hallazgo ha sido un indicador clásico de diagnóstico histológico de amiloidosis desde el siglo XIX6).
El tipo anteriormente llamado “distrofia corneal reticular tipo 2” es una manifestación ocular de la amiloidosis tipo gelsolina sistémica (GSN-AMYL, síndrome de Meretoja). En la clasificación IC3D actual, se clasifica como “amiloidosis familiar” y se trata de forma independiente de la LCD clásica4,10). Este síndrome, descrito por primera vez por Meretoja en Finlandia en 1969, es una enfermedad hereditaria caracterizada por opacidades reticulares corneales junto con neuropatía craneal progresiva, laxitud cutánea y síntomas sistémicos10,11). Dado que la diferenciación entre ambos es importante en la práctica clínica, en este artículo se describen ambos juntos.
Opacidades filamentosas de doble contorno en el área pupilar, erosiones epiteliales recurrentes
LCD IIIA (tipo variante)
TGFBI (5q31)
L527R, etc.
Después de los 40 años
Líneas reticulares gruesas en forma de cuerda en el estroma profundo, sin afectación epitelial
Tipo GSN (Meretoja)
GSN (9q34)
D187N, p.Glu580Lys2)
30 a 40 años
Líneas reticulares radiales en la periferia, amiloidosis sistémica
En Japón, la distrofia corneal asociada a TGFBI más frecuente es la tipo granular II (tipo Avellino, R124H), y la LCD1 es menos común. Sin embargo, dado que ambos tipos divergen por solo unos pocos pares de bases en el mismo gen TGFBI, se recomienda la prueba genética para la confirmación en casos con características clínicas superpuestas. No se ha informado la prevalencia exacta de LCD en general en Japón, pero es relativamente rara entre las distrofias corneales.
Q¿Cuál es la diferencia entre LCD1 y el síndrome de Meretoja?
A
La LCD1 es un depósito de amiloide limitado a la córnea causado por mutaciones en el gen TGFBI, que generalmente comienza en la zona pupilar en la segunda década de la vida y se asocia frecuentemente con erosiones epiteliales recurrentes. En contraste, el síndrome de Meretoja (anteriormente LCD2, tipo GSN) es una manifestación ocular de amiloidosis sistémica causada por mutaciones en el gen GSN (gelsolina), que comienza en la córnea periférica en la tercera o cuarta década, con transparencia central preservada durante mucho tiempo. El síndrome de Meretoja se acompaña de síntomas sistémicos como laxitud cutánea, facies de máscara, neuropatía periférica y arritmias cardíacas2,10). En la segunda edición de IC3D, el síndrome de Meretoja se clasifica de forma independiente de la distrofia corneal reticular4).
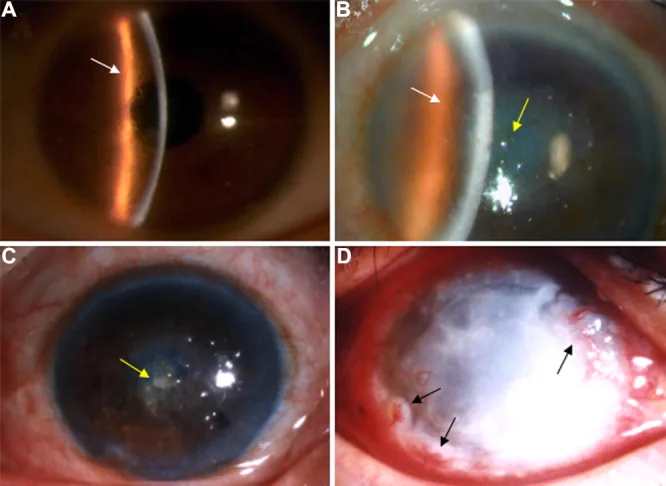
Liu Z, et al. An R124C mutation in TGFBI caused lattice corneal dystrophy type I with a variable phenotype in three Chinese families. Mol Vis. 2008. Figure 2. PMCID: PMC2443752. License: CC BY.
En la fotografía con lámpara de hendidura se observan líneas reticuladas ramificadas y opacidades de predominio central en el estroma corneal. Esta imagen muestra los hallazgos clínicos típicos de la distrofia corneal reticulada.
En LCD1, la mayoría de los pacientes son asintomáticos durante la infancia, con solo opacidades finas detectables mediante retroiluminación con lámpara de hendidura. Después de la segunda o tercera década de la vida, se producen erosiones corneales recurrentes (RCE) que causan dolor ocular agudo, fotofobia, lagrimeo y sensación de cuerpo extraño al despertar. Alrededor de los 30 años, aparecen opacidades blancas en el estroma anterior de la córnea central, y la agudeza visual disminuye progresivamente después de los 40 años.
En LCD IIIA (tipo variante), generalmente no se produce daño epitelial, y el síntoma principal es una disminución visual lenta después de los 40 años.
En el antiguo LCD2 (síndrome de Meretoja), los síntomas oculares aparecen entre los 30 y 40 años, pero el deterioro visual significativo a menudo se retrasa hasta los 60 años 11). Síntomas sistémicos como laxitud de la piel palpebral, facies de máscara, neuropatía craneal progresiva y arritmias cardíacas a menudo preceden o acompañan a los hallazgos oculares 2,10).
A continuación se muestran los hallazgos con lámpara de hendidura según el tipo de enfermedad.
LCD1 (tipo clásico)
Sitio inicial: Aparece como opacidades puntiformes y lineales finas en la capa de Bowman y el estroma anterior del área pupilar en ambos ojos.
Líneas reticuladas: Opacidades filamentosas o lineales con doble contorno se entrelazan formando opacidades reticulares o estrelladas.
Etapa avanzada: Se desarrollan opacidades blanquecinas en forma de yema de huevo o redondas en la córnea central.
Retroiluminación: Las líneas reticuladas finas y translúcidas que son difíciles de ver con iluminación directa se vuelven claramente visibles.
Tinción con fluoresceína: La reducción de la adhesión epitelial hace que la superficie se vea rugosa.
Erosión epitelial recurrente: Ocurre con frecuencia porque los depósitos se extienden a la membrana basal epitelial y la capa de Bowman.
LCD IIIA (tipo variante)
Líneas enrejadas: Líneas enrejadas gruesas y largas en el estroma medio a profundo, a veces con ramificaciones dendríticas. Observables incluso con iluminación directa.
Fenotipos: Hay tres patrones: (1) solo líneas enrejadas, (2) solo depósitos granulares pequeños, (3) una mezcla de ambos. En un mismo individuo, los ojos izquierdo y derecho pueden mostrar diferentes fenotipos, y también existen casos unilaterales.
Epitelio: Generalmente, no se produce daño epitelial.
Homocigotos: En homocigotos L527R, las líneas enrejadas son más gruesas y los depósitos granulares centrales son más grandes, pero la diferencia no es tan pronunciada como entre heterocigotos y homocigotos para R124H (tipo granular II).
Tipo GSN (Meretoja)
Líneas enrejadas: Un pequeño número de depósitos enrejados que carecen de delicadeza aparecen radialmente desde la periferia.
Transparencia central: La transparencia central se mantiene durante mucho tiempo después del inicio.
Erosión epitelial: Rara.
Hallazgos sistémicos: Se observan cambios faciales como facies en máscara, labios protuberantes con trastornos del movimiento, orejas caídas y blefarocalasia2).
En LCD1, algunos casos muestran una opacidad circular central particularmente intensa. Existe un informe de un heterocigoto R124C de 56 años que requirió trasplante de córnea debido a opacidad circular central.
Q¿Se puede diagnosticar LCD1 en niños?
A
La mayoría de los casos de LCD1 en la infancia son asintomáticos y las anomalías son difíciles de detectar solo con iluminación directa. La observación detallada con retroiluminación o iluminación de dispersión escleral del microscopio de lámpara de hendidura puede revelar opacidades puntiformes a lineales finas en el estroma superficial central. En niños con erosiones epiteliales corneales recurrentes, se debe considerar LCD1, y se recomienda una evaluación que incluya la historia familiar y el examen corneal de ambos padres. La prueba genética de TGFBI es útil para el diagnóstico definitivo.
Los genes causantes y las mutaciones representativas de la distrofia corneal enrejada se resumen a continuación.
Relacionado con TGFBI (LCD1, LCD IIIA, LCD IV)
Locus: 5q31 (gen TGFBI).
Herencia: Autosómica dominante.
Mutación común de LCD1: R124C (Arg124Cys) es la más frecuente5).
Mutación común de LCD IIIA: Se han reportado L527R (Leu527Arg) y otras. Existen casos homocigotos.
Mutación de novo: Se ha reportado un caso de mutación de novo de TGFBI L509P que presenta fenotipo de LCD IIIA1). Los padres no tenían la mutación, y un hijo la heredó1).
Rol de TGFBIp: Producido por el epitelio corneal, se distribuye en todo el espesor de la córnea y participa en el ensamblaje de fibras de colágeno en el estroma5).
Relacionado con GSN (síndrome de Meretoja, anteriormente LCD2)
Locus: 9q34 (gen GSN, gelsolina).
Herencia: Autosómica dominante.
Mutación clásica: D187N (tipo finlandés) es la más común; también se ha reportado p.Asp187Tyr10,11).
Mutación novedosa: p.Glu580Lys reportada en una familia eslovena se localiza en el límite del dominio G4-G5, causando repulsión electrostática debido a la sustitución de carga negativa a positiva2).
Características clínicas: Además de opacidades corneales en celosía, amiloidosis sistémica con laxitud cutánea, arritmia cardíaca, insuficiencia renal y neuropatía óptica2).
Al ser una enfermedad hereditaria, los antecedentes familiares son el factor de riesgo más importante. Sin embargo, pueden ocurrir mutaciones de novo en TGFBI, por lo que la ausencia de antecedentes familiares no la descarta1). El patrón de herencia es autosómico dominante; si uno de los padres es portador de la mutación, hay un 50% de probabilidad de transmisión a los hijos. No se observa diferencia de sexo, y las diferencias raciales no son claras para LCD1, pero se sabe que el síndrome de Meretoja se agrupa en familias finlandesas11).
La contribución de factores ambientales no es clara; la aparición y progresión de esta enfermedad están determinadas principalmente por el genotipo. Sin embargo, la frecuencia de erosiones epiteliales recurrentes puede exacerbarse por ambientes secos, uso de lentes de contacto o traumatismos. Las cirugías refractivas (LASIK, SMILE, etc.) pueden causar un empeoramiento rápido de las distrofias relacionadas con TGFBI, y se debe tener precaución en casos con antecedentes familiares durante el cribado preoperatorio5).
Para la diferenciación entre LCD1, tipos variantes y tipo GSN, se deben integrar los hallazgos de la lámpara de hendidura, los hallazgos histológicos y los hallazgos genéticos.
Examen clínico
Microscopía con lámpara de hendidura: Las líneas reticulares tempranas se pasan por alto fácilmente con iluminación directa. Utilice la retroiluminación para detectar opacidades finas contra el fondo de la pupila, y la iluminación indirecta para detectar líneas reticulares finas translúcidas.
Tinción con fluoresceína: En LCD1, la adhesión epitelial reducida resulta en una tinción rugosa. También es útil para evaluar la extensión de la erosión epitelial.
Microscopía confocal corneal: Permite observar los depósitos en el estroma a nivel celular.
Diagnóstico definitivo
Prueba genética: La detección de mutaciones en los genes TGFBI y GSN confirma el tipo de enfermedad. Incluso con el mismo fenotipo, diferentes mutaciones pueden alterar la tasa de recurrencia y progresión, lo que impacta directamente en la planificación del tratamiento.
Examen patológico: Se tiñe de rojo con rojo Congo y muestra birrefringencia verde manzana bajo luz polarizada, confirmando amiloide6).
Inmunohistoquímica: La diferenciación de tipos de enfermedad es posible usando anticuerpo anti-TGFBIp y anticuerpo anti-gelsolina.
Toma de antecedentes familiares: Dado que es un trastorno autosómico dominante, confirmar los hallazgos corneales en padres y hermanos respalda el diagnóstico.
Distrofia corneal granular tipo II (tipo Avellino, TGFBI R124H): La distrofia relacionada con TGFBI más común en Japón, que muestra una mezcla de depósitos granulares y líneas reticulares. La prueba genética es confiable para la diferenciación de LCD1.
Amiloidosis corneal secundaria: No hereditaria; el amiloide se deposita secundariamente debido a irritación crónica de la superficie ocular como triquiasis o queratocono. Los puntos de diferenciación incluyen ausencia de antecedentes familiares y presencia de enfermedad subyacente.
Distrofia corneal macular: Herencia autosómica recesiva por mutación del gen CHST6, con opacidad difusa en vidrio esmerilado y anomalías endoteliales.
Distrofia corneal gelatinosa en gotas: Herencia autosómica recesiva por mutación del gen TACSTD2, que presenta elevaciones gelatinosas de color blanco lechoso. Relativamente común en Japón.
Q¿Por qué es importante la prueba genética?
A
Incluso si la distrofia corneal reticular tiene fenotipos similares, las diferencias en los genes causantes y los sitios de mutación conducen a variaciones significativas en la tasa de progresión, frecuencia de recurrencia, opciones de tratamiento y presencia de complicaciones sistémicas. LCD1 con mutación TGFBI y el síndrome de Meretoja con mutación GSN tienen estrategias de tratamiento y necesidad de evaluación sistémica fundamentalmente diferentes2,10). Además, se han reportado mutaciones de novo, lo que hace imposible determinar el tipo de enfermedad basándose únicamente en los antecedentes familiares1). La prueba genética es esencial para el diagnóstico definitivo y la clasificación.
En la infancia hasta la juventud, cuando es asintomático o solo con opacidades finas, se realiza seguimiento. La progresión se evalúa mediante examen con lámpara de hendidura cada seis meses a un año.
Manejo de las erosiones epiteliales corneales recurrentes
Para las erosiones epiteliales recurrentes, un síntoma central de LCD1, la siguiente terapia conservadora es el primer paso.
Tratamiento agudo: Uso continuo de lentes de contacto blandas terapéuticas para proteger el epitelio corneal. Combinado con gotas antibacterianas para prevenir infección secundaria. Aplicar ungüento ocular para lubricación y protección epitelial.
Prevención de recurrencia: La administración de ungüento ocular antes de dormir es efectiva para suprimir la recurrencia de ataques de RCE. En ambientes secos, usar lágrimas artificiales o lubricantes también durante el día.
En LCD1, donde los depósitos de amiloide se encuentran principalmente en la córnea superficial, la queratectomía fototerapéutica (PTK) con láser excímero es la primera opción para casos con opacidad central severa o erosiones epiteliales corneales recurrentes7,8). Por lo general, no ocurre recurrencia temprana, pero la recurrencia con el tiempo es inevitable; la PTK se puede realizar hasta aproximadamente dos veces en el mismo ojo.
En heterocigotos, la recurrencia es lenta y pocos casos requieren retratamiento. En homocigotos, la recurrencia tiende a ocurrir más temprano que en heterocigotos. La tasa de recurrencia después de PTK aumenta con el tiempo, similar a otras distrofias relacionadas con TGFBI, y el seguimiento a largo plazo confirma algunos hallazgos de recurrencia en muchos casos8).
Como caso que demuestra la eficacia de PTK, en un caso de LCD IIIA debido a una mutación de novo en TGFBI L509P, se realizó PTK de 60 µm guiada por FD-OCT, y la mejor agudeza visual corregida (BCVA) mejoró de 20/400 a 20/501). No se observó disminución visual ni recurrencia significativa a los 45 meses postoperatorios1).
Según el Patrón de Práctica Preferida de la AAO para edema y opacidad corneal, la PTK para distrofias corneales granulares y enrejadas es una “opción razonable” y puede retrasar la transición a DALK o trasplante corneal de espesor total, pero existe riesgo de opacidad postoperatoria. Cuando se repite, se considera el uso de mitomicina C como medio para suprimir cicatrices recurrentes y depósitos estromales, y se advierte que el riesgo de ectasia corneal aumenta cuando la ablación supera el tercio anterior del estroma o cuando el lecho residual es menor de 250 µm7).
Se selecciona el trasplante de córnea para casos con recurrencia repetida o cuando la opacidad se extiende más allá del estroma medio. En LCD1, a menudo el trasplante de córnea no está indicado hasta después de los 40 años. En LCD, las células endoteliales corneales son generalmente normales, por lo que se selecciona el procedimiento quirúrgico según la profundidad de la opacidad.
Alta recuperación visual pero riesgo de rechazo y recurrencia
En los últimos años, la DALK se ha convertido en una nueva opción de primera línea ampliamente utilizada debido a la reducción del riesgo de rechazo y a resultados visuales comparables al trasplante de córnea de espesor total.
La recurrencia de LCD después del trasplante de córnea es inevitable, con tasas de recurrencia reportadas después del trasplante de córnea de espesor total del 17.8% a los 5 años, 26% a los 8 años y 56% a los 15 años 9). Las opacidades recurrentes generalmente se limitan a la capa superficial, por lo que pueden eliminarse con PTK, prolongando el intervalo hasta el retrasplante. Para LCD IIIA (tipo variante), a menudo no se requiere tratamiento a menos que la agudeza visual se vea afectada significativamente.
Q¿Qué tan efectiva es la PTK?
A
La PTK puede eliminar eficazmente los depósitos de amiloide superficiales, mejorando la agudeza visual y reduciendo las erosiones epiteliales recurrentes. En un caso de LCD IIIA, la mejor agudeza visual corregida mejoró de 20/400 a 20/50 después de PTK de 60 µm, sin recurrencia durante 45 meses 1). La recurrencia es lenta en heterocigotos pero temprana en homocigotos. Las lesiones profundas no pueden eliminarse con PTK, por lo que se requiere DALK o trasplante de córnea de espesor total para opacidades profundas 7).
Q¿Recurre la LCD después del trasplante de córnea?
A
La recurrencia de LCD después del trasplante de córnea es inevitable. Las tasas de recurrencia después del trasplante de córnea de espesor total se reportan como 17.8% a los 5 años, 26% a los 8 años y 56% a los 15 años 9). Sin embargo, las opacidades recurrentes generalmente se limitan a la capa superficial del injerto, por lo que pueden eliminarse con PTK, prolongando la supervivencia del injerto. La queratoplastia lamelar anterior profunda (DALK) tiene un menor riesgo de rechazo endotelial en comparación con el trasplante de córnea de espesor total y está ganando atención como una nueva opción de primera línea 7).
La patología central de la LCD1 es la acumulación anormal de TGFBIp (queratoepitelina, βig-h3). La TGFBIp es normalmente producida por el epitelio corneal y se distribuye por todo el espesor de la córnea, siendo una proteína estructural que participa en el ensamblaje de fibras de colágeno y la adhesión celular en el estroma 5). La proteína anormal producida por la mutación R124C sufre un plegamiento incorrecto y autoagregación, depositándose como fibrillas amiloides insolubles en la capa de Bowman y el estroma superficial. En etapas avanzadas, los depósitos se extienden al estroma profundo.
El depósito de amiloide altera las estructuras de adhesión epitelial de la córnea anterior, causando degeneración de las células basales del epitelio y degeneración de la capa epitelial acompañada de defectos en la membrana de Bowman. Esta alteración estructural constituye la base patológica de las erosiones epiteliales corneales recurrentes.
Diferenciación fenotípica según el genotipo de TGFBI
En el gen TGFBI, las diferencias en el sitio de mutación y el aminoácido sustituido determinan el fenotipo clínico. R124C causa LCD1, R124H causa distrofia corneal granular tipo II (tipo Avellino), y R124L causa distrofia corneal de Reis-Bücklers 5). El mecanismo molecular por el cual una diferencia de un solo aminoácido determina el material depositado (amiloide vs. hialino vs. ambos) y el sitio de depósito no se comprende completamente, pero se considera que el dominio de βig-h3 al que pertenece la mutación y su efecto sobre la estabilidad del plegamiento son factores clave.
En la LCD IIIA, mutaciones dominantes en capas profundas como L527R producen líneas reticulares gruesas en forma de cuerda y resultan en un tipo de inicio tardío sin afectación epitelial. La localización laminar de los depósitos puede explicarse por el gradiente de secreción y difusión de βig-h3 desde las células productoras (células basales del epitelio) hacia el estroma, y las diferencias en la estabilidad de plegamiento de la proteína mutante. Se cree que R124C favorece una vía desde intermediarios de plegamiento hacia la formación de fibrillas amiloides, acumulando amiloide alrededor de la capa de Bowman5). En cambio, la mutación L527R forma una proteína mal plegada relativamente estable que se deposita lentamente en capas estromales más profundas.
Amiloide corneal posterior y riesgo de cirugía intraocular
Convencionalmente, se pensaba que el depósito de amiloide en la LCD1 se limitaba a la córnea anterior (capa de Bowman a estroma superficial). Sin embargo, estudios patológicos recientes han demostrado que también existen depósitos de amiloide en la córnea posterior cerca de la membrana de Descemet3). El depósito de amiloide en la córnea posterior puede afectar la adhesión de la membrana de Descemet y contribuir a su desprendimiento durante la cirugía de cataratas 3). Se sugiere que un mecanismo similar al que el depósito de amiloide altera la adhesión epitelial en la córnea anterior también opera en la córnea posterior 3).
Mecanismo molecular de la amiloidosis tipo gelsolina
La gelsolina, la molécula causante del antiguo LCD2 (síndrome de Meretoja), existe tanto en el citoplasma como en el espacio extracelular y es una proteína involucrada en la motilidad celular, la división celular y la apoptosis a través de la unión a actina. La mutación clásica D187N, conocida como tipo finlandés, presenta depósitos corneales en forma de celosía y neuropatía craneal como fenotipo principal 11). La nueva mutación p.Glu580Lys reportada en una familia eslovena se localiza en el límite de los dominios G4-G5, y la sustitución de ácido glutámico con carga negativa por lisina con carga positiva provoca repulsión electrostática, reduciendo la conectividad y estabilidad entre dominios 2). La gelsolina mutante sufre un corte anómalo por furina y MT1-MMP en el plasma, liberando fragmentos precursores de amiloide de 8 kDa y 5 kDa. Estos se depositan en el estroma corneal, la piel, las paredes de los vasos sanguíneos, los nervios periféricos y los glomérulos renales, causando los síntomas multiorgánicos característicos del síndrome de Meretoja 2,11). Los depósitos corneales a menudo preceden a otros síntomas sistémicos, y los oftalmólogos pueden ser los primeros en diagnosticar esta enfermedad.
7. Investigación más reciente y perspectivas futuras
Se han reportado mutaciones de novo en el gen TGFBI que causan LCD 1). Incluso en casos sin antecedentes familiares, se debe considerar la posibilidad de mutaciones de novo y se recomienda la confirmación mediante pruebas genéticas 1). La mutación L509P es rara pero presenta un amplio espectro de fenotipos, desde similar a la distrofia corneal de Reis-Bücklers hasta similar a LCD IIIA 1).
En el gen GSN, además de las mutaciones convencionales p.Asp187Asn/Tyr, se ha reportado una nueva mutación p.Glu580Lys que se ha demostrado que causa amiloidosis sistémica con distrofia corneal reticular, laxitud cutánea, arritmia cardíaca, insuficiencia renal y neuropatía óptica2).
Lesiones corneales posteriores y manejo de cirugía intraocular
Estudios patológicos han demostrado que existen depósitos de amiloide en la córnea posterior de pacientes con LCD1 y pueden afectar la adhesión de la membrana de Descemet3). Se debe prestar atención al riesgo de desprendimiento de la membrana de Descemet durante cirugías intraoculares como la cirugía de cataratas.
Este hallazgo tiene implicaciones clínicas para la evaluación de indicaciones de cirugía de cataratas y la planificación quirúrgica en pacientes con LCD1.
Técnicas quirúrgicas asistidas por láser de femtosegundo
Se están desarrollando técnicas quirúrgicas más precisas como la queratectomía lamelar asistida por láser de femtosegundo (FLK) y la queratoplastia lamelar asistida por láser de femtosegundo (FALK) 12). Estas se están posicionando como opciones complementarias a la PTK convencional, ofreciendo una mejor suavidad de la superficie de resección y un control de profundidad altamente reproducible.
Dado que las mutaciones de TGFBI son mutaciones autosómicas dominantes de ganancia de función, se están investigando en fase preclínica ARNip específicos del alelo mutante, oligonucleótidos antisentido y el knockout alélico específico mediante CRISPR-Cas9. La córnea es ventajosa como órgano diana para la terapia génica porque permite la administración local y tiene privilegio inmunológico. Sin embargo, ninguna se ha aplicado clínicamente hasta la fecha, y todas requieren una futura verificación de seguridad y eficacia a largo plazo.
Inhibidores de la formación de amiloide y chaperonas moleculares
Se están investigando en fase de investigación básica compuestos de molécula pequeña dirigidos al proceso de agregación de TGFBIp y gelsolina mutante, chaperonas moleculares (como inductores de Hsp70) e inhibidores de la unión a fibrillas amiloides. Para la amiloidosis sistémica tipo gelsolina, algunos fármacos que inhiben la etapa de escisión de la gelsolina mutante en plasma se están evaluando en estudios preclínicos 2). En el futuro, se espera que estas terapias dirigidas molecularmente se conviertan en tratamientos fundamentales que reemplacen la resección física convencional (PTK y trasplante de córnea).
El análisis del proteoma corneal mediante espectrometría de masas ha sugerido que en LCD1, no solo TGFBIp sino también múltiples proteínas anormales pueden co-depositarse en los depósitos. Se está avanzando en dilucidar la contribución patogénica de estas proteínas co-depositadas para su futura aplicación clínica.
Ji YW, Ahn H, Shin KJ, Kim TI, Seo KY, Stulting RD, et al. De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. Journal of clinical medicine. 2022;11(11). doi:10.3390/jcm11113055. PMID:35683443; PMCID:PMC9181583.
Potrc M, Volk M, de Rosa M, Pizem J, Teran N, Jaklic H, Maver A, Drnovsek-Olup B, Bollati M, Vogelnik K, Hocevar A, Gornik A, Pfeifer V, Peterlin B, Hawlina M, Fakin A. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. Int J Mol Sci. 2021;22(3):1084.
Matsumoto A, Fukuoka H, Sotozono C. Descemet’s Membrane Detachment During Cataract Surgery in Lattice Corneal Dystrophy Type I: Histopathological Analysis of Posterior Corneal Involvement. Cureus. 2025;17(3):e81431. doi:10.7759/cureus.81431. PMID:40296953; PMCID:PMC12037203.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
Lisch W, Seitz B. The clinical landmarks of corneal dystrophies. Developments in ophthalmology. 2011;48:9-23. doi:10.1159/000324075. PMID:21540629.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern. San Francisco, CA: American Academy of Ophthalmology; 2024.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea. 2003;22(1):19-21.
Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969;1(4):314-324. PMID:4313418.
Kivelä T, Tarkkanen A, Frangione B, Ghiso J, Haltia M. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja’s syndrome. Invest Ophthalmol Vis Sci. 1994;35(10):3759-3769. PMID:8088963.
Steger B, Romano V, Biddolph S, Willoughby CE, Batterbury M, Kaye SB. Femtosecond laser-assisted lamellar keratectomy for corneal opacities secondary to anterior corneal dystrophies: an interventional case series. Cornea. 2016;35(1):6-13. PMID:26509759. doi:10.1097/ICO.0000000000000665.
Copia el texto del artículo y pégalo en el asistente de IA que prefieras.
Artículo copiado al portapapeles
Abre un asistente de IA abajo y pega el texto copiado en el chat.