LCD1(古典型)
初发部位:双眼瞳孔区的Bowman层至实质浅层出现微细点状、线状混浊。
格子状线:具有双重轮廓的丝状、线状混浊相互缠绕,形成网状、星状混浊。
进展期:中央部角膜出现卵黄形或圆形乳白色混浊。
反归照明法:直接照明下难以看清的半透明细格子状线清晰浮现。
荧光素染色:上皮粘附性降低导致表面粗糙。
复发性上皮糜烂:由于沉积物累及上皮基底细胞和Bowman膜,因此频繁发生。

格子状角膜营养不良(lattice corneal dystrophy, LCD)是一种遗传性角膜营养不良,淀粉样物质沉积于角膜基质,产生格子状线状混浊。该病历史悠久,早在1890年代就有记载,在IC3D(国际角膜营养不良分类委员会)第2版临床遗传学分类中,统一为LCD1及其变异型(原3型、3A型、1/3A型和4型)4)。
LCD1、颗粒状角膜营养不良、Reis-Bücklers角膜营养不良和Thiel-Behnke角膜营养不良构成一组疾病,称为“TGFBI相关营养不良”。致病基因TGFBI(转化生长因子β诱导基因)位于第5号染色体长臂(5q31),呈常染色体显性遗传。TGFBI蛋白(TGFBIp、kerato-epithelin、βig-h3)由角膜上皮细胞产生,分布于角膜全层。在角膜基质中,它参与胶原纤维的组装。即使同一基因的突变,由于突变位点和氨基酸替换的不同,沉积物(透明蛋白或淀粉样蛋白)和临床表现也有显著差异5)。
LCD1的代表性突变是TGFBI基因第124位精氨酸被半胱氨酸取代的R124C。在变异型LCD IIIA中,已报道有L527R等突变。
角膜中积累的异常蛋白经刚果红染色呈红色,在偏振光显微镜下呈现特有的苹果绿双折射,从而确认为淀粉样蛋白。这一发现自19世纪以来一直是淀粉样变性的经典组织诊断指标6)。
传统上称为“格子状角膜营养不良2型”的类型是全身性凝溶胶蛋白型淀粉样变性(GSN-AMYL,Meretoja综合征)的眼部表现,在当前的IC3D分类中被归类为“家族性淀粉样变性”,与经典LCD分开处理4,10)。该综合征于1969年由芬兰的Meretoja首次描述,是一种遗传性疾病,除角膜格子状混浊外,还伴有进行性脑神经病变、皮肤松弛和全身症状10,11)。由于临床上两者的鉴别很重要,本文一并描述。
| 病型 | 基因 | 代表性突变 | 发病年龄 | 主要表现 |
|---|---|---|---|---|
| LCD1(经典型) | TGFBI(5q31) | R124C | 10~20岁 | 瞳孔区双重轮廓丝状混浊,复发性上皮糜烂 |
| LCD IIIA(变异型) | TGFBI(5q31) | L527R 等 | 40岁以后 | 实质深层粗绳状格子线,无上皮病变 |
| GSN型(Meretoja) | GSN(9q34) | D187N、p.Glu580Lys2) | 30~40岁 | 周边部放射状格子线,全身性淀粉样变性 |
在日本,频率高的TGFBI相关角膜营养不良绝大多数是颗粒状II型(Avellino型、R124H),LCD1与之相比少见。但由于两者在相同的TGFBI基因上仅相差几个碱基而导致病型不同,因此在临床表现重叠的病例中,建议通过基因检测确诊。日本LCD整体的准确患病率尚未报告,但在角膜营养不良中属于相对罕见的类型。
LCD1是由TGFBI基因突变引起的局限于角膜的淀粉样沉积,1020岁从瞳孔区发病,常伴有复发性上皮糜烂。而Meretoja综合征(旧称LCD2、GSN型)是由GSN(凝溶胶蛋白)基因突变引起的全身性淀粉样变性的眼部表现,3040岁从周边角膜发病,中央透明性长期保持。Meretoja综合征伴有皮肤松弛、面具样面容、周围神经病变、心律失常等全身症状2,10)。在IC3D第2版中,Meretoja综合征被独立分类,不属于格子状角膜营养不良4)。
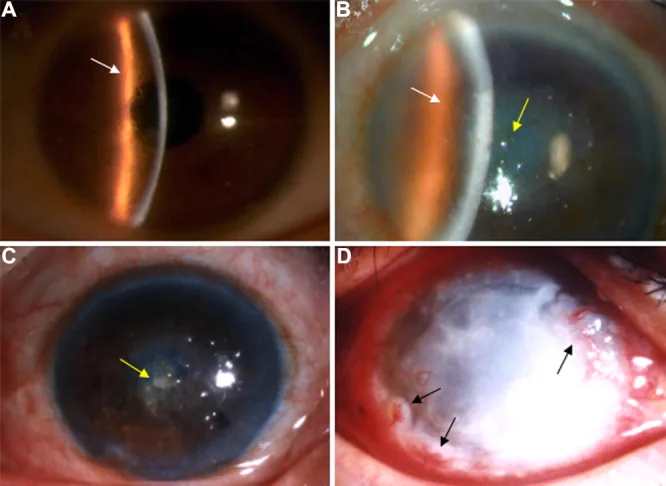
在LCD1中,儿童期多无症状,仅在裂隙灯显微镜的彻照法下可检测到细微混浊。10~20岁以后反复出现复发性角膜上皮糜烂(RCE),表现为晨起时的剧烈眼痛、畏光、流泪和异物感。30岁左右角膜中央部实质浅层出现白色混浊,40岁以后视力下降逐渐进展。
在LCD IIIA(变异型)中,通常不发生上皮损伤,主要症状为40岁以后缓慢的视力下降。
在旧LCD2(Meretoja综合征)中,眼部症状出现在30~40岁,但严重的视力障碍常延迟至60岁以后11)。眼睑皮肤松弛、面具样面容、进行性脑神经障碍、心脏心律失常等全身症状常先于或伴随眼部症状出现2,10)。
各病型的裂隙灯显微镜所见如下所示。
LCD1(古典型)
初发部位:双眼瞳孔区的Bowman层至实质浅层出现微细点状、线状混浊。
格子状线:具有双重轮廓的丝状、线状混浊相互缠绕,形成网状、星状混浊。
进展期:中央部角膜出现卵黄形或圆形乳白色混浊。
反归照明法:直接照明下难以看清的半透明细格子状线清晰浮现。
荧光素染色:上皮粘附性降低导致表面粗糙。
复发性上皮糜烂:由于沉积物累及上皮基底细胞和Bowman膜,因此频繁发生。
LCD IIIA(变异型)
格子状线:在实质中层至深层出现粗而长的格子状线,有时呈树枝状分支。直接照明下也可观察到。
表型:有三种模式:①仅有格子状线,②仅有小颗粒状沉积,③两者混合。同一个体左右眼可呈现不同表型,也有单眼病例。
上皮:通常不发生上皮损伤。
纯合子:L527R纯合子的格子状线更粗,中央颗粒状沉积更大,但不如R124H(颗粒状II型)杂合与纯合之间的差异显著。
GSN型(Meretoja)
格子状线:少数缺乏精细感的格子状沉积从周边部放射状出现。
中央透明性:发病后长期保持中央部透明。
上皮糜烂:罕见。
全身表现:出现面具样面容、伴有运动障碍的突出嘴唇、下垂的耳朵、眼睑皮肤松弛症等面部变化2)。
在LCD1中,有些病例中央部圆形混浊特别严重。有报道称一名56岁的R124C杂合子因中央圆形混浊需要角膜移植。
儿童期LCD1多无症状,直接照明下难以发现异常。使用裂隙灯显微镜的彻照法或反归照明法进行详细观察,可确认中央实质浅层的细微点状至线状混浊。对于反复发生角膜上皮糜烂的儿童,应考虑到LCD1,建议进行包括家族史询问和父母角膜检查在内的评估。TGFBI基因检测有助于确诊。
格子状角膜营养不良的致病基因和代表性突变总结如下。
TGFBI相关(LCD1、LCD IIIA、LCD IV)
基因座:5q31(TGFBI 基因)。
遗传方式:常染色体显性遗传。
LCD1 常见突变:R124C(Arg124Cys)最常见5)。
LCD IIIA 常见突变:已报道 L527R(Leu527Arg)等。存在纯合子病例。
新生突变:已报道 TGFBI L509P 的新生突变呈现 LCD IIIA 表型的病例1)。父母无突变,一个孩子遗传了该突变1)。
TGFBIp 的作用:由角膜上皮产生,分布于角膜全层,在基质中参与胶原纤维构建5)。
GSN相关(Meretoja综合征,旧称LCD2)
基因座:9q34(GSN 基因,凝溶胶蛋白)。
遗传方式:常染色体显性遗传。
经典突变:D187N(芬兰型)最常见,也有报道 p.Asp187Tyr10,11)。
新突变:斯洛文尼亚家族报道的 p.Glu580Lys 位于 G4-G5 结构域边界,因负电荷变为正电荷导致静电排斥2)。
临床表现:除角膜格子状混浊外,还伴有皮肤松弛、心律失常、肾损害和视神经病变的全身性淀粉样变性2)。
由于是遗传性疾病,家族史是最重要的风险因素。但 TGFBI 可能发生新生突变,因此无家族史不能排除1)。遗传方式为常染色体显性遗传,若父母一方携带突变,子女有 50% 的概率遗传。无性别差异,LCD1 种族差异不明显,但 Meretoja 综合征已知在芬兰有聚集性家族11)。
环境因素的贡献尚不明确,本病的发生和进展主要由基因型决定。但复发性上皮糜烂的频率可能因干燥环境、隐形眼镜佩戴或外伤而加重。屈光手术(LASIK、SMILE 等)可能导致 TGFBI 相关营养不良快速恶化,术前筛查中有家族史的病例需注意5)。
鉴别LCD1、变异型和GSN型需要综合裂隙灯所见、组织所见和基因所见。
临床检查
确诊
基因检测:检测TGFBI基因和GSN基因的突变可确定病型。即使表型相同,突变不同也会改变复发和进展的速度,因此直接关系到治疗计划。
病理检查:刚果红染色呈红色,偏振光显微镜下呈苹果绿双折射,可确认为淀粉样蛋白6)。
免疫组织化学:使用抗TGFBIp抗体和抗凝溶胶蛋白抗体可进行病型鉴别。
家族史询问:由于是常染色体显性遗传,确认父母和同胞的角膜所见有助于诊断。
即使格子状角膜营养不良表型相似,但致病基因和突变位点不同,其进展速度、复发频率、治疗选择和全身并发症的有无会有很大差异。TGFBI突变的LCD1和GSN突变的Meretoja综合征在治疗策略和全身检查的必要性上根本不同2,10)。此外,已有新生突变的报道,仅凭家族史无法确定疾病类型1),因此基因检测对于确诊和分型至关重要。
格子状角膜营养不良的治疗基于IC3D分类的以下阶梯式方法。
在儿童期至青年期,无症状或仅有细微混浊时进行随访观察。每半年至一年通过裂隙灯检查评估进展。
对于LCD1的核心症状——复发性上皮糜烂,以下保守治疗为首选。
在淀粉样沉积主要位于角膜表层的LCD1中,对于中央混浊严重或反复出现角膜上皮糜烂的病例,使用准分子激光的光治疗性角膜切除术(PTK)是首选7,8)。通常早期不会复发,但随时间推移复发不可避免;同一只眼可进行最多约两次PTK治疗。
杂合子中复发缓慢,需要再次治疗的病例很少。纯合子比杂合子更容易早期复发。与其他TGFBI相关营养不良一样,PTK后的复发率随时间增加,长期观察中许多病例会确认某种复发表现8)。
作为显示PTK有效性的病例,在由TGFBI L509P新生突变引起的LCD IIIA病例中,在FD-OCT引导下进行了60 µm的PTK,最佳矫正视力(BCVA)从20/400改善到20/501)。术后45个月时未观察到视力下降或显著复发1)。
AAO的角膜水肿和混浊优选实践模式指出,PTK对于颗粒状和格子状角膜营养不良是“合理的选择”,可能延迟向DALK或全层角膜移植的过渡,但存在术后混浊的风险。重复进行时,考虑联合使用丝裂霉素C作为抑制复发性瘢痕和实质沉积的手段,并警告当消融超过实质前三分之一或剩余床厚度小于250 µm时,角膜扩张的风险增加7)。
对于反复复发的病例,或混浊波及实质中深层时,选择角膜移植。在LCD1中,通常直到40岁以后才适合角膜移植。在LCD中,角膜内皮细胞通常正常,因此根据混浊深度选择术式。
| 术式 | 适应症 | 特点 |
|---|---|---|
| 表层角膜移植 | 浅层混浊 | 微创,排斥反应少 |
| 深板层角膜移植(DALK) | 中深层混浊 | 保留内皮,排斥风险低 |
| 穿透性角膜移植(PK) | 全层混浊 | 视力恢复好,但有排斥和复发风险 |
近年来,由于降低了排斥反应风险且视力结果与全层角膜移植相当,DALK 已广泛用作新的首选治疗方法。
角膜移植后 LCD 复发是不可避免的,全层角膜移植后的复发率报告为 5 年 17.8%、8 年 26%、15 年 56% 9)。复发性混浊通常局限于表层,因此可通过 PTK 去除,从而延长再次移植的间隔时间。对于 LCD IIIA(变异型),除非视力受到明显影响,否则通常不需要治疗。
LCD1 的核心病理是 TGFBIp(kerato-epithelin、βig-h3)的异常蓄积。TGFBIp 通常由角膜上皮产生,分布于角膜全层,在基质中参与胶原纤维构建和细胞粘附的结构蛋白5)。R124C 突变产生的异常蛋白发生错误折叠和自身聚集,以不溶性淀粉样原纤维的形式沉积在 Bowman 层和基质浅层。进展期时,沉积物向基质深层扩散。
淀粉样沉积导致前部角膜的上皮粘附结构发生变化,引起上皮基底细胞变性,以及伴有 Bowman 膜缺损的上皮层变性。这种结构破坏是复发性角膜上皮糜烂的病理基础。
TGFBI 基因中,突变位点和取代氨基酸的差异决定临床表型。R124C 导致 LCD1,R124H 导致颗粒状角膜营养不良 II 型(Avellino 型),R124L 导致 Reis-Bücklers 角膜营养不良5)。仅一个氨基酸的差异如何决定沉积物(淀粉样蛋白 vs 透明蛋白 vs 两者)和沉积部位的分子机制尚未完全阐明,但突变位点属于 βig-h3 的哪个结构域以及对折叠稳定性的影响被认为是关键因素。
在 LCD IIIA 中,L527R 等深层优势突变产生粗绳状格子线,形成不伴上皮损伤的晚发型。沉积物的层别定位可通过 βig-h3 的产生细胞(上皮基底细胞)向基质内的分泌和扩散梯度,以及突变蛋白折叠稳定性的差异来解释。R124C 被认为优先从折叠中间体走向淀粉样原纤维形成途径,在 Bowman 层周围蓄积淀粉样蛋白5)。而 L527R 突变形成相对稳定的错误折叠蛋白,缓慢沉积于更深的基质层。
传统认为 LCD1 的淀粉样沉积局限于前部角膜(Bowman 层至基质浅层)。但近年病理学研究显示,后部角膜近 Descemet 膜处也存在淀粉样沉积3)。后部角膜的淀粉样沉积可能影响 Descemet 膜的粘附,并在白内障手术中导致 Descemet 膜脱离3)。提示前部角膜中淀粉样沉积损害上皮粘附的类似机制也可能在后部起作用3)。
凝胶溶素(gelsolin)是旧LCD2(Meretoja综合征)的致病分子,存在于细胞质和细胞外,是一种通过肌动蛋白结合参与细胞运动、细胞分裂和凋亡的蛋白质。经典的D187N突变被称为芬兰型,主要表现为角膜格子状沉积和脑神经病变11)。在斯洛文尼亚家族中报道的新型p.Glu580Lys突变位于G4-G5结构域边界,带负电荷的谷氨酸被带正电荷的赖氨酸取代,产生静电排斥,从而降低结构域间的连接性和稳定性2)。突变型凝胶溶素在血浆中被弗林蛋白酶和MT1-MMP异常切割,释放出8 kDa和5 kDa的淀粉样前体片段。这些片段沉积在角膜基质、皮肤、血管壁、周围神经和肾小球中,引起Meretoja综合征特征性的多器官症状2,11)。角膜沉积通常先于其他全身症状,眼科医生可能是最早诊断该病的医生。
已有报道TGFBI基因的从头突变导致LCD的发生1)。即使没有家族史的病例,也应考虑从头突变的可能性,建议通过基因检测确认1)。L509P突变罕见,但呈现从Reis-Bücklers角膜营养不良样到LCD IIIA样的多种表型1)。
在GSN基因中,除了传统的p.Asp187Asn/Tyr突变外,还报道了新型p.Glu580Lys突变,该突变可导致伴有角膜格子状营养不良、皮肤松弛、心律失常、肾损伤和视神经病变的全身性淀粉样变性2)。
病理学研究表明,LCD1患者的后部角膜存在淀粉样沉积,可能影响Descemet膜的粘附3)。在白内障手术等内眼手术时,需注意Descemet膜脱离的风险。
这一发现对LCD1患者白内障手术适应症评估和手术方案制定具有临床意义。
飞秒激光辅助板层角膜切除术(FLK)和飞秒激光辅助板层角膜移植术(FALK)等更精确的手术技术正在开发中12)。这些技术通过提高切除面的平滑度和高度可重复的深度控制,正被定位为传统PTK的补充选择。
由于TGFBI突变是常染色体显性功能获得性突变,突变等位基因特异性siRNA、反义寡核苷酸以及CRISPR-Cas9介导的等位基因特异性敲除正在临床前研究阶段进行探讨。角膜因可局部给药且具有免疫特权,作为基因治疗的靶器官具有优势。但目前尚无临床应用的案例,均需要未来对长期安全性和有效性进行验证。
针对TGFBIp和突变凝溶胶蛋白聚集过程的小分子化合物、分子伴侣(如Hsp70诱导剂)以及淀粉样原纤维结合抑制剂正在基础研究阶段进行探讨。对于系统性凝溶胶蛋白型淀粉样变性,一些抑制血浆中突变凝溶胶蛋白裂解步骤的药物正在部分临床前试验中进行评估2)。未来,这类分子靶向治疗有望成为取代传统物理切除(PTK和角膜移植)的根本性治疗。
利用质谱法进行的角膜蛋白质组学分析表明,在LCD1的沉积物中,不仅TGFBIp,多种异常蛋白也可能共同沉积。为了未来的临床应用,正在阐明这些共沉积蛋白的病理贡献。