Loạn dưỡng giác mạc dạng lưới (lattice corneal dystrophy, LCD) là một chứng loạn dưỡng giác mạc di truyền, trong đó amyloid lắng đọng ở nhu mô giác mạc gây ra các vệt đục dạng lưới. Đây là bệnh được mô tả từ những năm 1890, và theo phân loại lâm sàng-di truyền của IC3D (Ủy ban Quốc tế về Phân loại Loạn dưỡng Giác mạc) phiên bản 2, được hợp nhất thành LCD1 và các biến thể của nó (các type 3, 3A, 1/3A, 4 trước đây)4).
LCD1, loạn dưỡng giác mạc dạng hạt, loạn dưỡng giác mạc Reis-Bücklers và loạn dưỡng giác mạc Thiel-Behnke tạo thành một nhóm bệnh gọi là “loạn dưỡng liên quan TGFBI”. Gen gây bệnh TGFBI (gen cảm ứng yếu tố tăng trưởng chuyển dạng beta) nằm trên nhánh dài nhiễm sắc thể số 5 (5q31), di truyền trội trên nhiễm sắc thể thường. Protein TGFBI (TGFBIp, kerato-epithelin, βig-h3) được sản xuất bởi biểu mô giác mạc và phân bố khắp các lớp giác mạc. Ở nhu mô giác mạc, nó tham gia vào cấu trúc sợi collagen. Ngay cả khi đột biến trên cùng một gen, sự khác biệt về vị trí đột biến và axit amin thay thế dẫn đến sự khác biệt lớn về chất lắng đọng (hyalin hay amyloid) và hình ảnh lâm sàng5).
Đột biến điển hình của LCD1 là R124C, trong đó arginine ở vị trí 124 của gen TGFBI được thay thế bằng cysteine. Ở thể biến thể LCD IIIA, các đột biến như L527R đã được báo cáo.
Protein bất thường tích tụ trong giác mạc bắt màu đỏ với nhuộm Congo red và thể hiện lưỡng chiết màu xanh táo đặc trưng dưới kính hiển vi phân cực, xác nhận đó là amyloid. Dấu hiệu này là chỉ số chẩn đoán mô học cổ điển của bệnh amyloidosis từ thế kỷ 196).
Thể trước đây gọi là ‘loạn dưỡng giác mạc dạng lưới type 2’ thực chất là biểu hiện ở mắt của bệnh amyloidosis toàn thân do gelsolin (GSN-AMYL, hội chứng Meretoja). Theo phân loại IC3D hiện tại, nó được xếp vào ‘bệnh amyloidosis gia đình’ và được xem xét độc lập với LCD cổ điển4,10). Hội chứng này được Meretoja người Phần Lan mô tả năm 1969, là bệnh di truyền bao gồm đục giác mạc dạng lưới kèm bệnh thần kinh sọ tiến triển, da chùng và các triệu chứng toàn thân10,11). Vì việc phân biệt hai bệnh này rất quan trọng trong thực hành lâm sàng, bài viết này trình bày cả hai.
Đục dạng sợi hai đường viền ở vùng đồng tử, trợt biểu mô tái phát
LCD IIIA (biến thể)
TGFBI (5q31)
L527R và các loại khác
Sau 40 tuổi
Các đường lưới dày như dây thừng ở lớp nhu mô sâu, không có tổn thương biểu mô
Loại GSN (Meretoja)
GSN (9q34)
D187N, p.Glu580Lys2)
30-40 tuổi
Các đường lưới xuyên tâm ở vùng ngoại vi, bệnh amyloidosis toàn thân
Ở Nhật Bản, loạn dưỡng giác mạc liên quan TGFBI phổ biến nhất là loại hạt II (Avellino, R124H), trong khi LCD1 ít gặp hơn. Tuy nhiên, vì cả hai đều do đột biến chỉ vài nucleotide trên cùng gen TGFBI, nên ở những bệnh nhân có biểu hiện lâm sàng chồng chéo, xét nghiệm di truyền được khuyến nghị để xác định chẩn đoán. Tỷ lệ hiện mắc chính xác của LCD nói chung tại Nhật Bản chưa được báo cáo, nhưng nó thuộc nhóm tương đối hiếm trong số các loạn dưỡng giác mạc.
QLCD1 và hội chứng Meretoja khác nhau như thế nào?
A
LCD1 là sự lắng đọng amyloid khu trú ở giác mạc do đột biến gen TGFBI, khởi phát ở vùng đồng tử từ 10-20 tuổi và thường kèm theo xói mòn biểu mô tái phát. Ngược lại, hội chứng Meretoja (trước đây gọi là LCD2, loại GSN) là biểu hiện ở mắt của bệnh amyloidosis toàn thân do đột biến gen GSN (gelsolin), khởi phát ở vùng ngoại vi giác mạc từ 30-40 tuổi và vùng trung tâm giữ được độ trong suốt lâu dài. Hội chứng Meretoja đi kèm với các triệu chứng toàn thân như da chùng, mặt nạ, bệnh thần kinh ngoại biên và rối loạn nhịp tim2,10). Trong phân loại IC3D phiên bản thứ hai, hội chứng Meretoja được phân loại độc lập với loạn dưỡng giác mạc dạng lưới4).
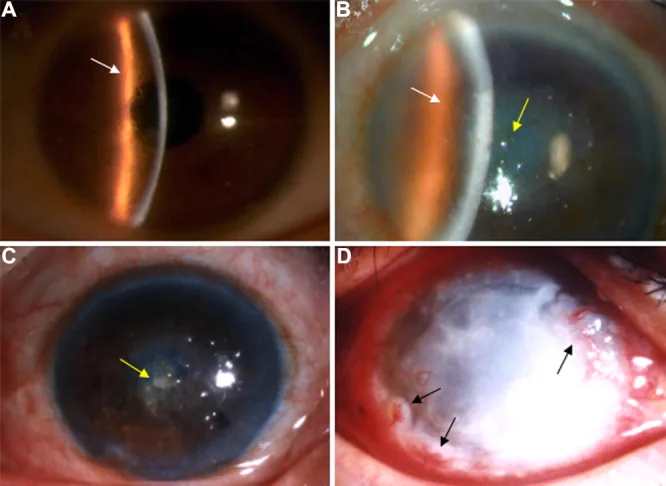
Liu Z, et al. An R124C mutation in TGFBI caused lattice corneal dystrophy type I with a variable phenotype in three Chinese families. Mol Vis. 2008. Figure 2. PMCID: PMC2443752. License: CC BY.
Trong ảnh đèn khe, có thể thấy các đường lưới phân nhánh ở nhu mô giác mạc và độ mờ chiếm ưu thế ở trung tâm. Đây là hình ảnh lâm sàng điển hình của loạn dưỡng giác mạc dạng lưới.
Trong LCD1, hầu hết bệnh nhân không có triệu chứng ở thời thơ ấu, chỉ có các đám mờ nhỏ được phát hiện bằng phương pháp chiếu xuyên qua của kính hiển vi đèn khe. Sau độ tuổi 10-20, tái phát xói mòn biểu mô giác mạc (recurrent corneal erosion, RCE) lặp đi lặp lại, gây đau mắt dữ dội, sợ ánh sáng, chảy nước mắt và cảm giác dị vật khi thức dậy. Khoảng 30 tuổi, các đám mờ trắng xuất hiện ở lớp nhu mô nông trung tâm giác mạc, và sau 40 tuổi, thị lực suy giảm tiến triển.
Trong LCD IIIA (thể biến thể), tổn thương biểu mô thường không xảy ra, và triệu chứng chính là suy giảm thị lực từ từ sau 40 tuổi.
Trong LCD2 cũ (hội chứng Meretoja), các triệu chứng về mắt xuất hiện ở độ tuổi 30-40, nhưng suy giảm thị lực nghiêm trọng thường bị trì hoãn đến 60 tuổi11). Các triệu chứng toàn thân như da mi mắt chùng, mặt nạ, bệnh thần kinh sọ tiến triển, rối loạn nhịp tim thường xuất hiện trước hoặc song song2,10).
Các dấu hiệu trên kính hiển vi đèn khe theo từng thể bệnh được trình bày dưới đây.
LCD1 (thể cổ điển)
Vị trí khởi phát: Xuất hiện dưới dạng các đám mờ dạng chấm nhỏ, dạng đường ở lớp Bowman đến lớp nhu mô nông ở vùng đồng tử của cả hai mắt.
Đường lưới: Các đám mờ dạng sợi, dạng đường có đường viền kép đan xen nhau, tạo thành các đám mờ dạng lưới hoặc hình sao.
Giai đoạn tiến triển: Xuất hiện đám mờ trắng đục hình trứng hoặc hình tròn ở trung tâm giác mạc.
Phương pháp chiếu ngược: Các đường lưới mảnh, trong mờ khó thấy dưới ánh sáng trực tiếp hiện lên rõ ràng.
Nhuộm fluorescein: Do giảm độ bám dính của biểu mô, bề mặt trở nên thô ráp.
Xói mòn biểu mô tái phát: Xảy ra thường xuyên do chất lắng đọng lan đến tế bào đáy biểu mô và màng Bowman.
LCD IIIA (thể biến thể)
Lưới dạng đường kẻ: Các đường kẻ dạng lưới dày và dài ở lớp nhu mô giữa đến sâu, đôi khi có dạng phân nhánh như cây. Có thể quan sát được dưới ánh sáng trực tiếp.
Kiểu hình: Có 3 dạng: ① chỉ có lưới dạng đường kẻ, ② chỉ có lắng đọng dạng hạt nhỏ, ③ hỗn hợp cả hai. Có thể có biểu hiện kiểu hình khác nhau giữa hai mắt trong cùng một cá thể hoặc trường hợp chỉ một mắt.
Biểu mô: Thông thường, không gây tổn thương biểu mô.
Đồng hợp tử: Ở đồng hợp tử L527R, các đường kẻ dạng lưới dày hơn và lắng đọng dạng hạt ở trung tâm lớn hơn, nhưng sự khác biệt không rõ rệt như giữa dị hợp tử và đồng hợp tử của R124H (dạng hạt loại II).
Loại GSN (Meretoja)
Lưới dạng đường kẻ: Một số ít lắng đọng dạng lưới kém tinh tế, xuất hiện tỏa tròn từ vùng ngoại vi.
Độ trong suốt trung tâm: Độ trong suốt của vùng trung tâm được duy trì trong thời gian dài sau khi khởi phát.
Xói mòn biểu mô: Hiếm gặp.
Biểu hiện toàn thân: Các thay đổi về ngoại hình như mặt nạ, môi nhô ra kèm rối loạn vận động, tai chảy xệ, da mi mắt chùng (blepharochalasis) 2).
Trong LCD1, một số trường hợp có độ đục hình tròn ở trung tâm đặc biệt nặng; có báo cáo về một bệnh nhân dị hợp tử R124C 56 tuổi có độ đục hình tròn trung tâm dẫn đến chỉ định ghép giác mạc.
QTrẻ em có thể được chẩn đoán LCD1 không?
A
LCD1 ở trẻ em thường không có triệu chứng, khó phát hiện bất thường chỉ bằng ánh sáng trực tiếp. Có thể xác nhận các đục dạng chấm đến dạng đường kẻ nhỏ ở lớp nhu mô nông trung tâm bằng cách quan sát chi tiết với kỹ thuật chiếu xuyên (transillumination) hoặc chiếu sáng phản xạ (retroillumination) của đèn khe. Ở trẻ em bị xói mòn biểu mô giác mạc tái phát, nên nghĩ đến LCD1, khuyến cáo khai thác tiền sử gia đình và kiểm tra giác mạc của cả cha mẹ. Xét nghiệm gen TGFBI hữu ích để chẩn đoán xác định.
Các gen gây bệnh và đột biến điển hình của loạn dưỡng giác mạc dạng lưới được tóm tắt dưới đây.
Liên quan TGFBI (LCD1, LCD IIIA, LCD IV)
Vị trí gen: 5q31 (gen TGFBI).
Kiểu di truyền: Di truyền trội trên nhiễm sắc thể thường.
Đột biến điển hình của LCD1: R124C (Arg124Cys) là phổ biến nhất5).
Đột biến điển hình của LCD IIIA: L527R (Leu527Arg) và các đột biến khác đã được báo cáo. Cũng có trường hợp đồng hợp tử.
Đột biến de novo: Đột biến de novo L509P của TGFBI đã được báo cáo ở một bệnh nhân có kiểu hình LCD IIIA1). Cả bố mẹ đều không có đột biến, nhưng đột biến đã được di truyền cho một trong những người con1).
Vai trò của TGFBIp: Được sản xuất bởi biểu mô giác mạc và phân bố khắp các lớp giác mạc; ở nhu mô, nó tham gia vào cấu trúc sợi collagen5).
Liên quan đến GSN (Hội chứng Meretoja, trước đây là LCD2)
Vị trí gen: 9q34 (gen GSN, gelsolin).
Kiểu di truyền: Di truyền trội trên nhiễm sắc thể thường.
Đột biến cổ điển: D187N (dạng Phần Lan) là phổ biến nhất; p.Asp187Tyr cũng đã được báo cáo10,11).
Đột biến mới: p.Glu580Lys được báo cáo trong một gia đình người Slovenia nằm ở ranh giới miền G4-G5, gây ra lực đẩy tĩnh điện do sự thay thế từ điện tích âm sang điện tích dương2).
Hình ảnh lâm sàng: Ngoài các đám mờ dạng lưới ở giác mạc, bệnh nhân còn biểu hiện bệnh amyloidosis toàn thân với da chùng, rối loạn nhịp tim, bệnh thận và bệnh thần kinh thị giác2).
Vì là bệnh di truyền, tiền sử gia đình là yếu tố nguy cơ quan trọng nhất. Tuy nhiên, đột biến de novo ở TGFBI có thể xảy ra, do đó không thể loại trừ bệnh chỉ dựa trên việc không có tiền sử gia đình1). Kiểu di truyền là trội trên nhiễm sắc thể thường; nếu một trong hai cha mẹ mang đột biến, con cái có 50% nguy cơ thừa hưởng đột biến. Không có sự khác biệt về giới tính; sự khác biệt về chủng tộc không rõ ràng ở LCD1, nhưng hội chứng Meretoja được biết đến với sự tập trung của các gia đình có nhiều người mắc ở Phần Lan11).
Vai trò của các yếu tố môi trường chưa rõ ràng; sự khởi phát và tiến triển của bệnh về cơ bản được xác định bởi kiểu gen. Tuy nhiên, tần suất xói mòn biểu mô tái phát có thể tăng lên trong môi trường khô, đeo kính áp tròng hoặc chấn thương. Phẫu thuật khúc xạ (LASIK, SMILE, v.v.) có thể gây ra sự xấu đi nhanh chóng của chứng loạn dưỡng giác mạc liên quan đến TGFBI; cần thận trọng ở những bệnh nhân có tiền sử gia đình khi sàng lọc trước phẫu thuật5).
Để phân biệt LCD1, thể biến thể và thể GSN, cần tổng hợp các dấu hiệu trên đèn khe, mô bệnh học và di truyền.
Xét nghiệm lâm sàng
Kính hiển vi đèn khe: Với ánh sáng trực tiếp dễ bỏ sót các đường lưới giai đoạn sớm. Phương pháp chiếu xuyên (retroillumination) phát hiện các vệt mờ mịn trên nền đồng tử, phương pháp phản chiếu (indirect illumination) phát hiện các đường lưới mảnh, trong mờ.
Nhuộm fluorescein: Ở LCD1, do giảm độ bám dính biểu mô, chất nhuộm bám dính trở nên thô ráp. Cũng hữu ích để đánh giá phạm vi trợt biểu mô.
Chụp cắt lớp quang học kết hợp vùng trước (OCT vùng trước): Có thể định lượng độ sâu phân lớp của lắng đọng. Đo độ sâu tổn thương bằng FD-OCT hữu ích để xác định độ sâu cắt trong PTK1).
Kính hiển vi đồng tiêugiác mạc: Có thể quan sát các lắng đọng trong nhu mô ở cấp độ tế bào.
Chẩn đoán xác định
Xét nghiệm di truyền: Phát hiện đột biến gen TGFBI và GSN để xác định thể bệnh. Cùng một kiểu hình nhưng đột biến khác nhau có thể thay đổi tốc độ tái phát và tiến triển, do đó liên quan trực tiếp đến kế hoạch điều trị.
Xét nghiệm mô bệnh học: Nhuộm Congo đỏ cho màu đỏ, dưới kính hiển vi phân cực cho thấy lưỡng chiết màu xanh táo, xác định là amyloid6).
Hóa mô miễn dịch: Có thể phân biệt thể bệnh bằng kháng thể kháng TGFBIp và kháng thể kháng gelsolin.
Khai thác tiền sử gia đình: Do di truyền trội trên nhiễm sắc thể thường, việc kiểm tra giác mạc của cha mẹ và anh chị em ruột hỗ trợ chẩn đoán.
Loạn dưỡng giác mạc dạng hạt type II (Avellino, TGFBI R124H): Là loạn dưỡng liên quan TGFBI phổ biến nhất ở Nhật Bản, biểu hiện hỗn hợp lắng đọng dạng hạt và đường lưới. Để phân biệt với LCD1, xét nghiệm di truyền là chắc chắn.
Bệnh amyloid giác mạc thứ phát: Không di truyền, amyloid lắng đọng thứ phát do kích thích bề mặt mắt mãn tính như lông mi mọc ngược hoặc giác mạc hình chóp. Điểm phân biệt là không có tiền sử gia đình và có bệnh nền.
Loạn dưỡng giác mạc dạng đốm: Di truyền lặn nhiễm sắc thể thường do đột biến gen CHST6, kèm đục dạng kính mờ lan tỏa và bất thường nội mô.
Loạn dưỡng giác mạc dạng giọt keo: Di truyền lặn nhiễm sắc thể thường do đột biến gen TACSTD2, biểu hiện các gờ nổi dạng keo trắng đục. Tương đối phổ biến ở Nhật Bản.
QTại sao xét nghiệm di truyền lại quan trọng?
A
Loạn dưỡng giác mạc dạng lưới dù có kiểu hình tương tự nhưng nếu gen gây bệnh và vị trí đột biến khác nhau thì tốc độ tiến triển, tần suất tái phát, lựa chọn điều trị và sự hiện diện của biến chứng toàn thân sẽ thay đổi đáng kể. LCD1 do đột biến TGFBI và hội chứng Meretoja do đột biến GSN có sự khác biệt cơ bản về phác đồ điều trị và nhu cầu thăm khám toàn thân2,10). Hơn nữa, đã có báo cáo về các trường hợp đột biến de novo không thể xác định thể bệnh chỉ dựa trên tiền sử gia đình1), do đó xét nghiệm di truyền là không thể thiếu để chẩn đoán xác định và phân loại thể bệnh.
Ở giai đoạn trẻ em đến thanh niên không có triệu chứng hoặc chỉ có đục nhẹ, tiến hành theo dõi. Đánh giá mức độ tiến triển bằng khám đèn khe mỗi 6 tháng đến 1 năm.
Đối với trợt biểu mô tái phát, triệu chứng cốt lõi của LCD1, các liệu pháp bảo tồn sau đây là bước đầu tiên.
Điều trị cơn cấp: Đeo kính áp tròng mềm điều trị liên tục để bảo vệ biểu mô giác mạc. Kết hợp nhỏ kháng sinh dự phòng nhiễm trùng thứ phát. Bôi thuốc mỡ tra mắt để bôi trơn và bảo vệ biểu mô.
Dự phòng tái phát: Bôi thuốc mỡ tra mắt trước khi ngủ có tác dụng ức chế tái phát cơn RCE. Trong môi trường khô, sử dụng nước mắt nhân tạo hoặc chất bôi trơn vào ban ngày.
Đối với LCD1 chủ yếu lắng đọng amyloid ở lớp nông giác mạc, khi đục trung tâm nặng hoặc trợt biểu mô tái phát nhiều lần, phẫu thuật cắt giác mạc điều trị bằng laser excimer (PTK) là lựa chọn đầu tiên7,8). Thông thường không tái phát sớm, nhưng tái phát theo thời gian là không thể tránh khỏi, và có thể thực hiện PTK tối đa khoảng 2 lần trên cùng một mắt.
Ở thể dị hợp tử, tái phát diễn ra chậm và ít trường hợp cần điều trị lại. Ở thể đồng hợp tử, có xu hướng tái phát sớm hơn so với thể dị hợp tử. Tỷ lệ tái phát sau PTK tăng dần theo thời gian, tương tự như các loạn dưỡng giác mạc liên quan TGFBI khác, và khi theo dõi lâu dài, hầu hết các trường hợp đều có dấu hiệu tái phát ở một mức độ nào đó8).
Một trường hợp minh họa hiệu quả của PTK là bệnh nhân LCD IIIA do đột biến de novo TGFBI L509P, được thực hiện PTK 60 µm dưới hướng dẫn FD-OCT, thị lực tốt nhất có điều chỉnh (BCVA) cải thiện từ 20/400 lên 20/501). Sau 45 tháng phẫu thuật, không ghi nhận suy giảm thị lực hay tái phát đáng kể1).
Theo Hướng dẫn Thực hành Ưu tiên về Phù và Đục giác mạc của AAO, PTK cho loạn dưỡng giác mạc dạng hạt và dạng lưới là ‘lựa chọn hợp lý’ và có thể trì hoãn việc chuyển sang DALK hoặc ghép giác mạc toàn bộ, nhưng có nguy cơ đục giác mạc sau phẫu thuật. Khi thực hiện lặp lại, việc kết hợp mitomycin C được xem xét như một biện pháp ức chế sẹo tái phát và lắng đọng nhu mô. Cảnh báo rằng nguy cơ giãn giác mạc tăng cao nếu vùng cắt sâu quá 1/3 trước nhu mô hoặc lớp đệm còn lại dưới 250 µm7).
Đối với các trường hợp tái phát nhiều lần hoặc đục lan đến lớp trung bì sâu hơn, nên chọn ghép giác mạc. Trong LCD1, thường không có chỉ định ghép giác mạc cho đến khi bệnh nhân trên 40 tuổi. Vì tế bào nội mô giác mạc trong LCD thường bình thường, nên lựa chọn phương pháp phẫu thuật dựa trên độ sâu của đục.
Phục hồi thị lực cao nhưng có nguy cơ thải ghép và tái phát
Trong những năm gần đây, DALK đã được sử dụng rộng rãi như một lựa chọn đầu tay mới nhờ giảm nguy cơ thải ghép và kết quả thị lực tương đương với ghép giác mạc toàn bộ.
Tái phát LCD sau ghép giác mạc là không thể tránh khỏi, tỷ lệ tái phát sau ghép toàn bộ được báo cáo là 17,8% sau 5 năm, 26% sau 8 năm và 56% sau 15 năm9). Đục tái phát thường khu trú ở lớp bề mặt, do đó có thể loại bỏ bằng PTK và kéo dài thời gian đến khi ghép lại. Đối với LCD IIIA (thể biến thể), thường không cần điều trị trừ khi ảnh hưởng nhiều đến thị lực.
QPTK có hiệu quả đến mức nào?
A
PTK có thể loại bỏ hiệu quả lắng đọng amyloid ở lớp bề mặt, cải thiện thị lực và giảm xói mòn biểu mô tái phát. Trong một trường hợp LCD IIIA, thị lực tốt nhất đã cải thiện từ 20/400 lên 20/50 sau PTK 60 µm và không tái phát trong 45 tháng1). Ở thể dị hợp tử, tái phát diễn ra chậm, nhưng ở thể đồng hợp tử, tái phát sớm xảy ra. Các tổn thương sâu không thể loại bỏ bằng PTK, do đó cần DALK hoặc ghép giác mạc toàn bộ cho các đục sâu7).
QBệnh có tái phát sau ghép giác mạc không?
A
Tái phát LCD sau ghép giác mạc là không thể tránh khỏi. Tỷ lệ tái phát sau ghép giác mạc toàn bộ được báo cáo là 17,8% sau 5 năm, 26% sau 8 năm và 56% sau 15 năm9). Tuy nhiên, đục tái phát thường khu trú ở lớp bề mặt của mảnh ghép, do đó có thể loại bỏ bằng PTK và kéo dài tuổi thọ của mảnh ghép. Ghép giác mạc lớp sâu (DALK) có nguy cơ thải ghép nội mô thấp hơn so với ghép toàn bộ và đang được chú ý như một lựa chọn đầu tay mới7).
Trọng tâm bệnh lý của LCD1 là sự tích tụ bất thường của TGFBIp (kerato-epithelin, βig-h3). TGFBIp bình thường được biểu mô giác mạc sản xuất và phân bố khắp các lớp giác mạc, là protein cấu trúc tham gia vào việc xây dựng sợi collagen và kết dính tế bào ở nhu mô5). Protein bất thường được tạo ra do đột biến R124C gây ra sự gấp cuộn sai và tự kết tụ, lắng đọng dưới dạng sợi amyloid không hòa tan ở lớp Bowman và nhu mô nông. Ở giai đoạn tiến triển, sự lắng đọng lan rộng vào sâu trong nhu mô.
Sự lắng đọng amyloid gây ra những thay đổi trong cấu trúc bám dính biểu mô ở giác mạc trước, dẫn đến thoái hóa tế bào đáy biểu mô và thoái hóa lớp biểu mô kèm theo khiếm khuyết màng Bowman. Sự phá vỡ cấu trúc này là nền tảng bệnh lý của xói mòn biểu mô giác mạc tái phát.
Ở gen TGFBI, sự khác biệt về vị trí đột biến và axit amin thay thế quyết định hình ảnh lâm sàng. R124C gây ra LCD1, R124H gây ra loạn dưỡng giác mạc dạng hạt type II (type Avellino), R124L gây ra loạn dưỡng giác mạc Reis-Bücklers5). Cơ chế phân tử mà chỉ một axit amin khác biệt quyết định chất lắng đọng (amyloid vs hyalin vs cả hai) và vị trí lắng đọng vẫn chưa được làm sáng tỏ hoàn toàn, nhưng vị trí đột biến thuộc miền nào của βig-h3 và ảnh hưởng đến độ ổn định gấp cuộn được cho là chìa khóa.
Trong LCD IIIA, các đột biến ưu thế ở lớp sâu như L527R tạo ra các đường dạng lưới dày giống dây thừng, trở thành type khởi phát muộn không kèm tổn thương biểu mô. Sự khu trú theo lớp của chất lắng đọng có thể được giải thích bởi gradient tiết và khuếch tán từ tế bào sản xuất βig-h3 (tế bào đáy biểu mô) vào nhu mô, cùng với sự khác biệt về độ ổn định gấp cuộn của protein đột biến. R124C ưu tiên con đường từ trung gian gấp cuộn đến hình thành sợi amyloid, tích tụ amyloid quanh lớp Bowman5). Trong khi đó, đột biến L527R tạo thành protein gấp cuộn sai tương đối ổn định và lắng đọng chậm ở lớp nhu mô sâu hơn.
Amyloid giác mạc sau và nguy cơ phẫu thuật nội nhãn
Trước đây, lắng đọng amyloid trong LCD1 được cho là giới hạn ở giác mạc trước (lớp Bowman đến nhu mô nông). Tuy nhiên, các nghiên cứu bệnh lý gần đây cho thấy lắng đọng amyloid cũng tồn tại ở giác mạc sau gần màng Descemet3). Lắng đọng amyloid ở giác mạc sau có thể ảnh hưởng đến sự bám dính của màng Descemet và góp phần gây bong màng Descemet trong phẫu thuật đục thủy tinh thể3). Cơ chế tương tự như lắng đọng amyloid ở giác mạc trước làm tổn thương sự bám dính biểu mô được cho là cũng tác động ở giác mạc sau3).
Gelsolin, phân tử gây ra hội chứng LCD2 cũ (hội chứng Meretoja), tồn tại cả trong tế bào chất và ngoại bào, là một protein tham gia vào vận động tế bào, phân chia tế bào và apoptosis thông qua liên kết actin. Đột biến cổ điển D187N, được gọi là type Phần Lan, biểu hiện kiểu hình với lắng đọng dạng lưới ở giác mạc và bệnh lý thần kinh sọ não 11). Đột biến mới p.Glu580Lys được báo cáo trong một gia đình người Slovenia nằm ở ranh giới miền G4-G5, gây ra sự thay thế axit glutamic mang điện tích âm bằng lysin mang điện tích dương, dẫn đến lực đẩy tĩnh điện và làm giảm tính liên kết và ổn định giữa các miền 2). Gelsolin đột biến bị phân cắt bất thường bởi furin và MT1-MMP trong huyết tương, giải phóng các mảnh tiền thân amyloid 8 kDa và 5 kDa. Các mảnh này lắng đọng ở nền giác mạc, da, thành mạch, dây thần kinh ngoại biên và cầu thận, gây ra các triệu chứng đa cơ quan đặc trưng của hội chứng Meretoja 2,11). Lắng đọng giác mạc thường xuất hiện trước các triệu chứng toàn thân khác, và bác sĩ nhãn khoa có thể là người đầu tiên chẩn đoán bệnh này.
Sự xuất hiện của LCD do đột biến de novo trên gen TGFBI đã được báo cáo 1). Ngay cả ở những bệnh nhân không có tiền sử gia đình, cũng cần xem xét khả năng đột biến de novo và khuyến cáo xác nhận bằng xét nghiệm di truyền 1). Đột biến L509P hiếm gặp nhưng biểu hiện kiểu hình đa dạng từ loạn dưỡng giác mạc Reis-Bücklers đến dạng LCD IIIA 1).
Trên gen GSN, ngoài đột biến p.Asp187Asn/Tyr đã biết, một đột biến mới p.Glu580Lys đã được báo cáo, gây ra bệnh amyloidosis toàn thân với loạn dưỡng giác mạc dạng lưới, da chùng, rối loạn nhịp tim, bệnh thận và bệnh thần kinh thị giác2).
Tổn thương giác mạc sau và quản lý phẫu thuật nội nhãn
Đã được chứng minh về mặt bệnh lý rằng có sự lắng đọng amyloid ở giác mạc sau của bệnh nhân LCD1, có thể ảnh hưởng đến sự bám dính của màng Descemet3). Cần chú ý đến nguy cơ bong màng Descemet trong các phẫu thuật nội nhãn như phẫu thuật đục thủy tinh thể.
Phát hiện này có ý nghĩa lâm sàng trong việc đánh giá chỉ định phẫu thuật đục thủy tinh thể và lập kế hoạch phẫu thuật cho bệnh nhân LCD1.
Các kỹ thuật phẫu thuật chính xác hơn như cắt giác mạc lớp có hỗ trợ laser femtosecond (femtosecond laser-assisted lamellar keratectomy, FLK) và ghép giác mạc lớp có hỗ trợ laser femtosecond (femtosecond laser-assisted lamellar keratoplasty, FALK) đang được phát triển 12). Chúng đang được coi là lựa chọn bổ sung cho PTK truyền thống nhờ cải thiện độ mịn của bề mặt cắt và kiểm soát độ sâu có độ tái lập cao.
Vì đột biến TGFBI là đột biến tăng chức năng trội trên nhiễm sắc thể thường, các phương pháp như siRNA đặc hiệu alen đột biến, oligonucleotide antisense và knockout đặc hiệu alen bằng CRISPR-Cas9 đang được nghiên cứu ở giai đoạn tiền lâm sàng. Giác mạc là cơ quan đích thuận lợi cho liệu pháp gen vì có thể dùng thuốc tại chỗ và có đặc quyền miễn dịch. Tuy nhiên, hiện chưa có phương pháp nào được ứng dụng lâm sàng, và tất cả đều cần được kiểm chứng về tính an toàn và hiệu quả lâu dài trong tương lai.
Thuốc ức chế hình thành amyloid và chaperone phân tử
Các hợp chất phân tử nhỏ nhắm vào quá trình kết tụ của TGFBIp và gelsolin đột biến, chaperone phân tử (như thuốc cảm ứng Hsp70) và thuốc ức chế gắn kết sợi amyloid đang được nghiên cứu ở giai đoạn cơ bản. Đối với bệnh amyloidosis dạng gelsolin toàn thân, một số thuốc ức chế giai đoạn cắt gelsolin đột biến trong huyết tương đang được đánh giá trong các thử nghiệm tiền lâm sàng 2). Trong tương lai, các liệu pháp nhắm đích phân tử này được kỳ vọng sẽ thay thế các phương pháp cắt bỏ vật lý truyền thống (PTK, ghép giác mạc) như một liệu pháp triệt để.
Phân tích proteome giác mạc bằng khối phổ cho thấy trong các chất lắng đọng của LCD1, không chỉ TGFBIp mà còn có thể có nhiều protein bất thường khác cùng lắng đọng. Hướng tới ứng dụng lâm sàng trong tương lai, vai trò bệnh lý của các protein đồng lắng đọng này đang được làm sáng tỏ.
Ji YW, Ahn H, Shin KJ, Kim TI, Seo KY, Stulting RD, et al. De Novo L509P Mutation of the TGFBI Gene Associated with Slit-Lamp Findings of Lattice Corneal Dystrophy Type IIIA. Journal of clinical medicine. 2022;11(11). doi:10.3390/jcm11113055. PMID:35683443; PMCID:PMC9181583.
Potrc M, Volk M, de Rosa M, Pizem J, Teran N, Jaklic H, Maver A, Drnovsek-Olup B, Bollati M, Vogelnik K, Hocevar A, Gornik A, Pfeifer V, Peterlin B, Hawlina M, Fakin A. Clinical and Histopathological Features of Gelsolin Amyloidosis Associated with a Novel GSN Variant p.Glu580Lys. Int J Mol Sci. 2021;22(3):1084.
Matsumoto A, Fukuoka H, Sotozono C. Descemet’s Membrane Detachment During Cataract Surgery in Lattice Corneal Dystrophy Type I: Histopathological Analysis of Posterior Corneal Involvement. Cureus. 2025;17(3):e81431. doi:10.7759/cureus.81431. PMID:40296953; PMCID:PMC12037203.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
Lisch W, Seitz B. The clinical landmarks of corneal dystrophies. Developments in ophthalmology. 2011;48:9-23. doi:10.1159/000324075. PMID:21540629.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern. San Francisco, CA: American Academy of Ophthalmology; 2024.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Marcon AS, Cohen EJ, Rapuano CJ, Laibson PR. Recurrence of corneal stromal dystrophies after penetrating keratoplasty. Cornea. 2003;22(1):19-21.
Meretoja J. Familial systemic paramyloidosis with lattice dystrophy of the cornea, progressive cranial neuropathy, skin changes and various internal symptoms. A previously unrecognized heritable syndrome. Ann Clin Res. 1969;1(4):314-324. PMID:4313418.
Kivelä T, Tarkkanen A, Frangione B, Ghiso J, Haltia M. Ocular amyloid deposition in familial amyloidosis, Finnish: an analysis of native and variant gelsolin in Meretoja’s syndrome. Invest Ophthalmol Vis Sci. 1994;35(10):3759-3769. PMID:8088963.
Steger B, Romano V, Biddolph S, Willoughby CE, Batterbury M, Kaye SB. Femtosecond laser-assisted lamellar keratectomy for corneal opacities secondary to anterior corneal dystrophies: an interventional case series. Cornea. 2016;35(1):6-13. PMID:26509759. doi:10.1097/ICO.0000000000000665.
Sao chép toàn bộ bài viết và dán vào trợ lý AI bạn muốn dùng.
Đã sao chép bài viết vào clipboard
Mở một trợ lý AI bên dưới và dán nội dung đã sao chép vào ô chat.