Dạng quả dâu tằm
dạng dâu tằm điển hình: Dạng đặc trưng nhất.
Trung tâm giác mạc: Các tổn thương nhô cao màu trắng xám tụ lại giống quả dâu tằm.
Amyloid dưới biểu mô: Các khối giống keo màu trắng sữa bán trong suốt tăng dần từ trung tâm ra ngoại vi.

Loạn dưỡng giác mạc dạng giọt keo (gelatinous drop-like corneal dystrophy: GDLD) là một bệnh giác mạc di truyền, trong đó amyloid lắng đọng dưới biểu mô giác mạc, gây giảm thị lực đáng kể ở cả hai mắt.
Bệnh này được báo cáo lần đầu tiên vào năm 1914 bởi Nakaizumi, và vào năm 1932, Kiyosawa đặt tên là “thoái hóa giác mạc dạng giọt keo”, và từ đó được gọi như vậy. Trong phân loại IC3D (Ủy ban Quốc tế về Phân loại Loạn dưỡng Giác mạc), nó được xếp vào loạn dưỡng biểu mô, viết tắt là GDLD.
Gen gây bệnh được xác định vào năm 1999 bởi Tsujikawa và cộng sự, đó là gen TACSTD2 (tumor-associated calcium signal transducer 2), một gen đơn exon nằm trên nhiễm sắc thể 1p324).
Năm 2019, bệnh được công nhận là bệnh hiếm gặp cụ thể “Loạn dưỡng giác mạc dạng giọt keo” và được hỗ trợ chi phí y tế 2). Tiêu chuẩn chẩn đoán và phân loại mức độ nặng đã được xây dựng bởi Dự án Nghiên cứu Chính sách Bệnh Nan y của Bộ Y tế, Lao động và Phúc lợi 2).
GDLD đã được báo cáo trên toàn thế giới, nhưng phổ biến hơn ở Nhật Bản. Hầu như không có ca bệnh ở châu Âu và Bắc Mỹ. Hơn 20 đột biến đã được báo cáo trên gen TACSTD2, cho thấy tính không đồng nhất di truyền. Ở Nhật Bản, đột biến Q118X nằm trên 1p32 là đột biến sáng lập phổ biến, chiếm hơn 80% nhiễm sắc thể gây bệnh ở bệnh nhân Nhật Bản.
Thường khởi phát trước 20 tuổi. Bệnh nhân phàn nàn về các triệu chứng sau từ khi còn nhỏ.
Khi tuổi tác tăng lên, số lượng và kích thước của các lắng đọng amyloid tăng lên. Các lắng đọng trở thành màu trắng xám đến vàng, và cuối cùng bao phủ hầu hết giác mạc tập trung ở khe mi 2). Xâm nhập mạch máu từ ngoại vi, giảm thị lực rõ rệt và đau mắt xảy ra, cùng với các vấn đề thẩm mỹ, làm giảm đáng kể chất lượng cuộc sống của bệnh nhân.
Độ đục giác mạc được phân loại thành 4 loại dựa trên hình thái. Các loại này có thể được phân biệt bằng cách khám đoạn trước với đèn khe 2,3).
Dạng quả dâu tằm
dạng dâu tằm điển hình: Dạng đặc trưng nhất.
Trung tâm giác mạc: Các tổn thương nhô cao màu trắng xám tụ lại giống quả dâu tằm.
Amyloid dưới biểu mô: Các khối giống keo màu trắng sữa bán trong suốt tăng dần từ trung tâm ra ngoại vi.
Dạng loạn dưỡng giác mạc dạng dải
dạng loạn dưỡng giác mạc dạng dải: Có thể thấy ở giai đoạn sớm.
Giữa khe mi: Độ đục ở lớp nông. Biểu hiện tương tự loạn dưỡng giác mạc dạng dải.
Tổn thương kết mạc: Cũng có thể thấy tổn thương ở kết mạc.
Dạng quả kim quất
dạng quả kim quất: Thường gặp ở giai đoạn tiến triển.
Lắng đọng vàng trắng lan tỏa: Toàn bộ giác mạc chuyển màu vàng và trông như quả kim quất.
Xâm nhập mạch máu: Có thể kèm theo tân mạch nông.
Loại đục mô đệm
loại đục mô đệm: giai đoạn tiến triển hơn.
Lan rộng vào mô đệm: Tổn thương lan đến mô đệm giác mạc.
Xâm nhập mạch máu: Kèm theo xâm nhập mạch máu vào các khối u dạng keo màu trắng sữa đến vàng.
Ide và cộng sự đã báo cáo phổ lâm sàng chi tiết của 34 trường hợp tại Nhật Bản và chỉ ra rằng ngay cả với cùng một đột biến gen TACSTD2 (đồng hợp tử Q118X), bốn kiểu hình có thể cùng tồn tại 3).
Ngoài ra, có các dấu hiệu đặc trưng sau:
GDLD gây ra bởi các đột biến mất chức năng trong gen TACSTD2 4).
Do bất thường gen TACSTD2, sự định vị nội bào bình thường của Claudin 1 và Claudin 7, các protein cấu thành điểm tiếp xúc chặt ở biểu mô giác mạc, bị mất, làm giảm chức năng hàng rào biểu mô 5). Kết quả là, các protein như lactoferrin từ nước mắt xâm nhập vào giác mạc, hình thành các sợi amyloid và lắng đọng dưới biểu mô. Nakatsuka và cộng sự đã chứng minh bằng sinh học phân tử thông qua phân tích các gia đình Nhật Bản rằng TACSTD2 cần thiết cho sự định vị claudin bình thường, và làm rõ rằng sinh lý bệnh của GDLD là do rối loạn chức năng điểm tiếp xúc chặt 5).
Tiêu chuẩn chẩn đoán GDLD đã được xây dựng bởi nhóm nghiên cứu của Dự án Nghiên cứu Chính sách Bệnh Nan y của Bộ Y tế, Lao động và Phúc lợi “Xây dựng Hướng dẫn Thực hành Lâm sàng cho các Bệnh Nan y Phần trước Mắt và Phổ biến, Giáo dục” 2). Nếu được phân loại là Xác định (Definite) theo các tiêu chuẩn này, bệnh sẽ trở thành đối tượng của bệnh hiếm được chỉ định.
A. Triệu chứng (có bất kỳ triệu chứng nào)
B. Kết quả Xét nghiệm
C. Chẩn đoán phân biệt: Loại trừ bệnh amyloidosis giác mạc thứ phát và bệnh lý giác mạc do giọt khí hậu
D. Biến chứng ngoài mắt: Không có
E. Xét nghiệm di truyền: Có bất thường ở gen TACSTD2
Điều kiện Chắc chắn được đáp ứng nếu một trong các điều sau được thỏa mãn2).
B1 (lắng đọng dạng quả dâu tằm) là một dấu hiệu rất đặc trưng, và trong các trường hợp điển hình, chẩn đoán không khó. Trong các trường hợp không điển hình, chẩn đoán được thực hiện bằng cách kết hợp A-C và xét nghiệm di truyền (E)2).
Mức độ nặng được phân loại thành độ I-IV dựa trên thị lực đã chỉnh của mắt tốt hơn2).
| Mức độ nặng | Tiêu chuẩn | Hỗ trợ chi phí y tế |
|---|---|---|
| Độ I | Chỉ một mắt bị ảnh hưởng, mắt kia bình thường | × |
| Độ II | Cả hai mắt bị ảnh hưởng, thị lực điều chỉnh của mắt tốt hơn ≥ 0,3 | × |
| Độ III | Cả hai mắt bị ảnh hưởng, thị lực điều chỉnh của mắt tốt hơn ≥ 0,1 và < 0,3 | ○ |
| Độ IV | Cả hai mắt bị ảnh hưởng, thị lực điều chỉnh của mắt tốt hơn < 0,1 | ○ |
Khi được chẩn đoán là Definite, bệnh nhân sẽ được coi là mắc bệnh hiếm được chỉ định và có thể nhận hỗ trợ chi phí y tế nếu mức độ nghiêm trọng từ độ III trở lên2). Nếu có hẹp thị trường (thị trường trung tâm còn lại ≤ 20 độ với thị kích thước Goldmann I/4) ở mắt tốt hơn do glôcôm thứ phát hoặc các nguyên nhân khác, mức độ nghiêm trọng sẽ tăng lên một bậc.
Xét nghiệm gen TACSTD2 đã được bảo hiểm y tế chi trả từ năm 2020 như “Xét nghiệm gen loạn dưỡng giác mạc (D006-20)”. Tuy nhiên, cơ sở y tế cần có hệ thống cho phép thực hiện xét nghiệm và được công nhận. TACSTD2 là gen đơn exon nên dễ dàng tìm kiếm, và hơn 80% bệnh nhân Nhật Bản có đột biến sáng lập Q118X, do đó đặc biệt hữu ích trong chẩn đoán các trường hợp không điển hình 2).
Điều trị GDLD được lựa chọn dựa trên mức độ đục và mức độ suy giảm thị lực. Vì là bệnh di truyền, thách thức lớn nhất là tỷ lệ tái phát rất cao với bất kỳ phương pháp điều trị nào 2). Không hiếm trường hợp biến chứng do ghép giác mạc nhiều lần và glôcôm thứ phát dẫn đến mù lòa.
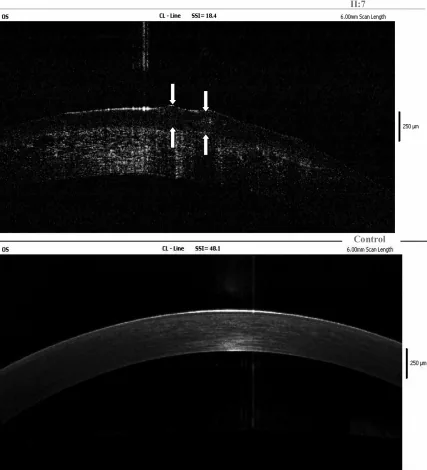
Đeo kính áp tròng mềm điều trị liên tục được xem xét như liệu pháp bảo tồn và hỗ trợ 6). Maeno và cộng sự năm 2020 trong một nghiên cứu quan sát tiến cứu trên bệnh nhân GDLD đã chỉ ra rằng đeo kính áp tròng điều trị ức chế đáng kể sự tái phát của các tổn thương dạng gờ gelatin màu xám đến vàng 7). Cũng được khuyến cáo để phòng ngừa tái phát sau phẫu thuật.
PTK
Cắt bỏ giác mạc nông bằng laser excimer điều trị: Lựa chọn đầu tiên cho đục giác mạc nông.
Chỉ định: Tổn thương dạng gờ gelatin nông giai đoạn sớm đến trung bình. Kết hợp với nạo thủ công. Kết quả dài hạn 8,9).
Ghép giác mạc
Ghép vùng rìa giác mạc
Ghép tế bào gốc vùng rìa và tạo biểu mô giác mạc: Kết hợp với ghép giác mạc.
Mục đích: Che phủ bề mặt nhãn cầu bằng biểu mô giác mạc có nguồn gốc từ mảnh ghép và ngăn chặn sự xâm nhập trở lại của biểu mô vật chủ 10,11).
Kết quả dài hạn của PTK đã được báo cáo từ Nhật Bản. Ōura và cộng sự đã chỉ ra kết quả dài hạn của PTK trên các trường hợp GDLD và báo cáo tính hữu ích của nó trong việc kéo dài thời gian đến tái phát 8). Hieda và cộng sự trong một nghiên cứu đa trung tâm tại Nhật Bản đã phân tích chi tiết thời điểm tái phát và kết quả lâm sàng sau PTK 9).
Kết hợp ghép tế bào gốc vùng rìa (LSCT) là một phương pháp tiếp cận có nguồn gốc từ Nhật Bản được đánh giá cao trên toàn cầu. Shimazaki và cộng sự năm 2002 đã báo cáo hiệu quả của ghép giác mạc kết hợp LSCT cho GDLD, cho thấy có thể kéo dài thời gian đến tái phát bằng cách ngăn chặn sự xâm nhập trở lại của tế bào biểu mô vật chủ 10). Sau đó, Movahedan và cộng sự cũng báo cáo phương pháp tiếp cận tương tự 11).
Để che phủ bề mặt nhãn cầu bằng biểu mô giác mạc từ mảnh ghép, biểu mô giác mạc của vật chủ được loại bỏ, sau đó tiến hành ghép vùng rìa. Sau phẫu thuật, tiếp tục đeo kính áp tròng điều trị liên tục, đồng thời làm chậm tái phát.
Trong những năm gần đây, việc sử dụng giác mạc nhân tạo (Boston type I Kpro) cũng đang được xem xét. Về mặt lý thuyết, có thể tránh tái lắng đọng amyloid vì không qua biểu mô giác mạc của vật chủ, nhưng có nguy cơ biến chứng sau phẫu thuật như nhiễm trùng và màng sau giác mạc nhân tạo.
Trong GDLD, tỷ lệ tái phát sau ghép giác mạc là rất cao. Khoảng 97% tái phát trong vòng 4 năm sau ghép giác mạc xuyên thấu (PKP) đã được báo cáo. Nguyên nhân chính là sự thay thế tế bào biểu mô của người nhận bằng biểu mô mảnh ghép. Là biện pháp xuất phát từ Nhật Bản, việc trì hoãn tái phát được thực hiện bằng cách kết hợp ghép tế bào gốc vùng rìa giác mạc 10) và đeo kính áp tròng điều trị liên tục 7). Trong quản lý dài hạn, mục tiêu là duy trì chức năng thị giác và kéo dài khoảng cách giữa các lần phẫu thuật, với giả định tái phát.
Gen TACSTD2, gen gây bệnh GDLD, là gen đơn exon nằm trên nhiễm sắc thể 1p32. Nó được xác định là gen gây bệnh vào năm 1999 bởi Tsujikawa và cộng sự thông qua phân tích liên kết trên các gia đình Nhật Bản 4). Protein TACSTD2 đóng vai trò thiết yếu trong việc duy trì chức năng hàng rào của biểu mô giác mạc.
Khi xảy ra đột biến mất chức năng ở gen TACSTD2, sự định vị nội bào bình thường của các protein mối nối chặt Claudin 1 và Claudin 7 bị suy giảm. Nakatsukasa và cộng sự đã chỉ ra, thông qua phân tích tế bào biểu mô giác mạc nuôi cấy và các gia đình Nhật Bản, rằng mất chức năng TACSTD2 dẫn đến mất định vị Claudin tại mối nối đỉnh-bên và giảm chức năng hàng rào biểu mô 5). Hơn nữa, vào năm 2011, họ đã báo cáo các đột biến mới của TACSTD2 ở 3 gia đình và sự định vị nội bào bất thường của chúng 12).
Do chức năng hàng rào biểu mô suy giảm, các protein như lactoferrin từ nước mắt xâm nhập vào giác mạc. Lactoferrin xâm nhập hình thành các sợi amyloid và lắng đọng dưới biểu mô giác mạc. Các lắng đọng amyloid có chứa lactoferrin, nhưng bệnh này không phải do bất thường gen lactoferrin.
Về mặt mô học, độ đục màu trắng đục dưới biểu mô bắt màu đỏ cam với nhuộm Congo red, và thể hiện lưỡng chiết màu xanh táo dưới kính hiển vi phân cực. Dưới kính hiển vi điện tử, các điểm nối chặt của biểu mô được thay thế bằng các khoảng trống điện tử. Các chất lắng đọng cũng xâm nhập vào các lớp mô giác mạc, gây thoái hóa các sợi collagen và proteoglycan.
Hơn 20 đột biến đã được báo cáo trên gen TACSTD212). Tại Nhật Bản, đột biến Q118X (đột biến vô nghĩa, null chức năng) là đột biến sáng lập chiếm hơn 80% nhiễm sắc thể gây bệnh2). Bệnh thường biểu hiện ở thể đồng hợp tử, nhưng cũng xảy ra ở thể dị hợp tử phức hợp do hôn nhân giữa các gia đình khác nhau. Điều thú vị là, ngay cả ở những người đồng hợp tử Q118X giống hệt nhau, cả bốn phân nhóm (dâu tằm, dạng dải, quả quất và đục nhu mô) đều được quan sát thấy pha trộn3).
Bệnh thoái hóa dạng tinh bột giác mạc được phân loại thành nguyên phát hoặc thứ phát, toàn thân hoặc khu trú. GDLD được phân loại là bệnh thoái hóa dạng tinh bột khu trú nguyên phát cùng với loạn dưỡng giác mạc dạng lưới. Thoái hóa dạng tinh bột khu trú thứ phát xảy ra do lông quặm, giác mạc hình chóp, chấn thương hoặc đeo kính áp tròng kéo dài, và cần được chẩn đoán phân biệt.
GDLD là một bệnh hiếm gặp, và có sự thiếu hụt bác sĩ có kinh nghiệm lâm sàng tại từng cơ sở, cũng như thiếu các phương pháp chẩn đoán và điều trị tiêu chuẩn. Nhóm Nghiên cứu Khảo sát Dịch tễ học các Bệnh Giác mạc Hiếm gặp và Khó chữa của Bộ Y tế, Lao động và Phúc lợi, và nhóm Nghiên cứu Phát triển và Phổ biến Hướng dẫn Lâm sàng cho các Bệnh Khó chữa Phân đoạn Trước đã xây dựng các tiêu chuẩn chẩn đoán và phân loại mức độ nghiêm trọng2). Năm 2019, “Loạn dưỡng giác mạc dạng giọt keo” đã được công nhận là bệnh hiếm được chỉ định, và hiện đang được xây dựng hướng dẫn lâm sàng dựa trên Minds2).
Về lâu dài, tái phát và điều trị nhiều lần là vấn đề. Kết hợp kính áp tròng điều trị và ghép vùng rìa có thể làm chậm tái phát và kéo dài khoảng cách giữa các lần phẫu thuật, cải thiện tiên lượng suốt đời6, 7, 10).
Maeno và cộng sự đã báo cáo một ca lâm sàng không điển hình có lắng đọng amyloid tái phát chỉ ở một mắt, cho thấy sự đa dạng kiểu hình của GDLD13). Xét nghiệm gen TACSTD2 đóng vai trò quyết định trong chẩn đoán các ca không điển hình này2).
Trong nghiên cứu cơ bản, dự kiến sẽ làm sáng tỏ chi tiết động lực học phân tử của các mối nối chặt chẽ ở hạ lưu TACSTD2 và phát triển các liệu pháp nhắm mục tiêu ổn định Claudin. Về mặt lâm sàng, việc mở rộng chỉ định của các phương pháp tái tạo như giác mạc nhân tạo, ghép tấm biểu mô giác mạc và biểu mô giác mạc có nguồn gốc từ tế bào iPS đang được xem xét.