A Distrofia Corneana em Gotas Gelatinosas (gelatinous drop-like corneal dystrophy: GDLD) é uma doença corneana hereditária na qual amiloide se deposita abaixo do epitélio corneano, causando diminuição acentuada da acuidade visual em ambos os olhos.
Esta doença foi relatada pela primeira vez em 1914 por Nakaizumi, e em 1932 Kiyosawa a nomeou “degeneração corneana em gotas gelatinosas”, sendo chamada assim desde então. Na classificação IC3D (International Committee for Classification of Corneal Dystrophies), é classificada como distrofia epitelial, com a abreviatura GDLD.
O gene causador foi identificado em 1999 por Tsujikawa et al., sendo o gene TACSTD2 (tumor-associated calcium signal transducer 2), um gene de éxon único localizado no cromossomo 1p324).
Prevalência: Raro globalmente, mas relatado com relativa frequência no Japão 1). Acredita-se que a diminuição dos casamentos consanguíneos tenha reduzido a frequência de ocorrência 2)
Diferenças regionais: Relativamente comum no Japão, quase não relatado na Europa e América do Norte
Mutação Q118X: Mutação fundadora em pacientes japoneses, representando mais de 80% dos cromossomos patogênicos 2)
Idade de início: Frequentemente se manifesta antes dos 20 anos
Em 2019, foi designada como doença rara específica “Distrofia Corneana Gotosa Gelatinosa” e tornou-se elegível para subsídios médicos 2). Os critérios diagnósticos e a classificação de gravidade foram elaborados pelo Projeto de Pesquisa de Políticas para Doenças Intratáveis do Ministério da Saúde, Trabalho e Bem-Estar 2).
QA GDLD ocorre fora do Japão?
A
A GDLD foi relatada em todo o mundo, mas é mais comum no Japão. Quase não há casos na Europa e América do Norte. Mais de 20 mutações foram relatadas no gene TACSTD2, indicando heterogeneidade genética. No Japão, a mutação Q118X localizada em 1p32 é uma mutação fundadora frequente, representando mais de 80% dos cromossomos patogênicos em pacientes japoneses.
Frequentemente se manifesta antes dos 20 anos. Os pacientes queixam-se dos seguintes sintomas desde a infância.
Fotofobia: Sintoma proeminente desde o início
Sensação de corpo estranho: Devido às elevações gelatinosas na superfície da córnea
Lacrimejamento: Acompanhando sintomas de irritação
Baixa acuidade visual: Piora gradualmente com a progressão dos depósitos amiloides. Torna-se acentuada após a idade adulta
Com o envelhecimento, o número e o tamanho dos depósitos de amiloide aumentam. Os depósitos tornam-se esbranquiçados a amarelados e, eventualmente, cobrem a maior parte da córnea centrada na fenda palpebral 2). Ocorre invasão vascular a partir da periferia, diminuição acentuada da acuidade visual e dor ocular, além de problemas estéticos, reduzindo significativamente a qualidade de vida do paciente.
Achados Clínicos (Achados Confirmados pelo Médico no Exame)
A opacidade corneana é classificada em 4 tipos de acordo com a morfologia. Esses tipos podem ser diferenciados pelo exame do segmento anterior com lâmpada de fenda2,3).
Tipo Amora
tipo amora típico: O tipo mais característico.
Centro da córnea: Lesões elevadas esbranquiçadas agrupadas que se assemelham a uma amora.
Amiloide subepitelial: Elevações coloides semitransparentes branco-leitosas que aumentam do centro para a periferia.
Tipo Ceratopatia em Banda
tipo ceratopatia em banda: Pode ser observado em estágios iniciais.
Entre as pálpebras: Opacidade nas camadas superficiais. Apresenta achados semelhantes à ceratopatia em banda.
Lesões conjuntivais: Lesões também podem ser encontradas na conjuntiva.
Tipo Kumquat
tipo kumquat: Comum em casos avançados.
Depósitos amarelo-esbranquiçados difusos: Toda a córnea torna-se amarelada e assume aparência de kumquat.
Invasão vascular: Pode ser acompanhada de neovascularização superficial.
Tipo de opacidade estromal
tipo de opacidade estromal: estágio mais avançado.
Extensão para o estroma: A lesão se estende ao estroma corneano.
Invasão vascular: Acompanhada de invasão vascular em elevações gelatinosas de cor branco-leitosa a amarelada.
Ide et al. relataram o espectro clínico detalhado de 34 casos no Japão e mostraram que, mesmo com a mesma mutação no gene TACSTD2 (homozigoto Q118X), quatro fenótipos podem se misturar 3).
Além disso, existem os seguintes achados característicos:
Coloração tardia com fluoresceína: Apesar de não haver anormalidade epitelial corneana, devido ao aumento da permeabilidade por formação imperfeita das junções oclusivas, observa-se fluorescência minutos após a instilação de fluoresceína2)
Afinaçao epitelial: O epitélio corneano é fino nas áreas com elevações gelatinosas
Invasão vascular: Observa-se invasão vascular superficial na periferia da córnea
Devido à anormalidade no gene TACSTD2, a localização intracelular normal da Claudina 1 e Claudina 7, proteínas constituintes das junções oclusivas no epitélio corneano, é perdida, reduzindo a função de barreira epitelial 5). Como resultado, proteínas como a lactoferrina das lágrimas invadem a córnea, formam fibras amiloides e depositam-se sob o epitélio. Nakatsuka et al. demonstraram por biologia molecular, por meio da análise de famílias japonesas, que o TACSTD2 é essencial para a localização normal da claudina, e esclareceram que a fisiopatologia da GDLD é devida à disfunção das junções oclusivas 5).
História familiar: Por ser uma herança autossômica recessiva, há risco de desenvolver a doença se ambos os pais forem portadores
Ascendência japonesa: A mutação sem sentido Q118X é uma mutação fundadora no Japão, representando mais de 80% dos cromossomos causadores da doença 2)
Casamento consanguíneo: Geralmente, os pais do probando são casados entre parentes. No entanto, a doença também pode ocorrer em heterozigotos compostos de casamentos entre famílias diferentes
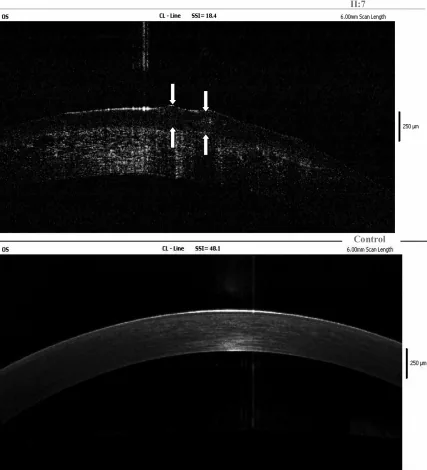
Yang Jing, Chun Liu, Liya Wang A novel TACSTD2 mutation identified in two Chinese brothers with gelatinous drop-like corneal dystrophy 2009 Aug 14 Mol Vis. 2009 Aug 14; 15:1580-1588 Figure 5. PMCID: PMC2728569. License: CC BY.
Achados de OCT de domínio de Fourier da córnea esquerda do probando mostram depósitos de amiloide no epitélio e no estroma superficial, e as setas indicam a localização das lesões em gotas gelatinosas. Isso corresponde aos depósitos de amiloide discutidos na seção “4. Diagnóstico e Métodos de Exame”.
Critérios Diagnósticos (Grupo de Estudo do Ministério da Saúde, Trabalho e Bem-Estar)
Os critérios diagnósticos para GDLD foram estabelecidos pelo grupo de estudo do Projeto de Pesquisa de Políticas para Doenças Intratáveis do Ministério da Saúde, Trabalho e Bem-Estar “Desenvolvimento de Diretrizes de Prática Clínica para Doenças Intratáveis do Segmento Anterior do Olho e sua Disseminação e Educação” 2). Se classificado como Definitivo (Definite) de acordo com esses critérios, torna-se elegível como doença rara designada.
A. Sintomas (apresentar qualquer um)
Diminuição da acuidade visual
Fotofobia
Sensação de corpo estranho
Lacrimejamento
B. Achados de Exame
Presença de aglomerados de depósitos amiloides subepiteliais da córnea (em forma de amora) de cor branco-acinzentada e elevados na região central da córnea até a fissura palpebral em ambos os olhos
Presença de coloração tardia observada minutos após a coloração com fluoresceína, apesar da ausência de anormalidades epiteliais da córnea
Presença de invasão vascular superficial na periferia da córnea
C. Diagnóstico diferencial: Excluir amiloidose corneana secundária e ceratopatia climática em gotículas
D. Complicações extraoculares: Nenhuma
E. Exame genético: Presença de anormalidade no gene TACSTD2
A condição Definite é atendida se um dos seguintes for satisfeito2).
Caso que atenda D, qualquer um de A, e B1, e possa excluir as doenças a serem diferenciadas em C
Caso que atenda D, qualquer um de A, e B2 ou B3, e atenda E, e possa excluir as doenças a serem diferenciadas em C
B1 (depósitos em forma de amora) é um achado muito característico, e em casos típicos o diagnóstico não é difícil. Em casos atípicos, o diagnóstico é feito combinando A-C e exame genético (E)2).
Classificação de gravidade (de acordo com o anúncio de doenças raras designadas)
A gravidade é classificada em graus I-IV com base na acuidade visual corrigida do olho melhor2).
Gravidade
Critério
Apoio financeiro médico
Grau I
Apenas um olho afetado, o outro olho é saudável
×
Grau II
Ambos os olhos afetados, acuidade visual corrigida do melhor olho ≥ 0,3
×
Grau III
Ambos os olhos afetados, acuidade visual corrigida do melhor olho ≥ 0,1 e < 0,3
○
Grau IV
Ambos os olhos afetados, acuidade visual corrigida do melhor olho < 0,1
○
Quando diagnosticado como Definite, torna-se elegível como doença rara designada, e pode receber auxílio para custos médicos se a gravidade for Grau III ou superior2). Se houver estreitamento do campo visual (campo visual central remanescente ≤ 20 graus com o alvo Goldmann I/4) no melhor olho devido a glaucoma secundário, etc., a gravidade é elevada em um grau.
Exame com lâmpada de fenda: Observar lesões elevadas acinzentadas do centro da córnea até a área da fissura palpebral, e distinguir 4 tipos (tipo amora, tipo em faixa, tipo kumquat, tipo opacidade parenquimatosa)
Teste de permeabilidade à fluoresceína (coloração tardia): Devido à disfunção das junções oclusivas, o corante penetra rapidamente no tecido corneano 2)
Teste genético TACSTD2: Foi coberto pelo seguro saúde desde 2020 como teste genético para distrofia corneana (D006-20) 2). TACSTD2 é um gene de éxon único, facilitando a pesquisa. Particularmente útil no diagnóstico de casos atípicos
Exame histológico (corte de córnea): Cora-se em vermelho alaranjado com vermelho Congo e mostra birrefringência verde-maçã sob microscópio de polarização, confirmando amiloide
Amiloidose corneana secundária: Depósito de amiloide devido a estímulo crônico como triquíase, entrópio, ápice do ceratocone ou uso de lentes de contato rígidas. Ausência de história familiar e presença de inflamação crônica da superfície ocular são pontos diferenciais. Pode apresentar elevações gelatinosas ou aspecto reticulado, necessitando de exame histológico para confirmação
Ceratopatia climática em gotículas: Mais comum em homens acima de 40 anos. Encontrada em regiões desérticas ou muito frias, causada por radiação UV e ressecamento. Apresenta lesões corneanas elevadas amareladas a branco-acinzentadas
Degeneração corneana em faixa: Depósito de sais de cálcio no subepitélio. Inicia-se na periferia às 3 e 9 horas e progride para o centro
Distrofia corneana reticular tipo I: Causada pela mutação R124C no gene TGFBI, herança autossômica dominante. Apresenta opacidades fibrosas ramificadas no estroma corneano
Distrofia corneana macular: Causada por alteração no gene CHST6, herança autossômica recessiva. Opacidade difusa em vidro fosco
QO teste genético é coberto pelo seguro?
A
O teste genético TACSTD2 é coberto pelo seguro saúde desde 2020 como “Teste genético para distrofia corneana (D006-20)”. No entanto, a instituição precisa ter um sistema que permita a realização do teste e obter acreditação. TACSTD2 é um gene de éxon único, facilitando a pesquisa, e mais de 80% dos pacientes japoneses têm a mutação fundadora Q118X, sendo particularmente útil no diagnóstico de casos atípicos 2).
O tratamento da GDLD é escolhido de acordo com a extensão da opacidade e o grau de comprometimento visual. Por ser uma doença hereditária, o maior desafio é a altíssima taxa de recorrência com qualquer tratamento 2). Não é raro que complicações de transplantes repetidos de córnea e glaucoma secundário levem à cegueira.
Lágrimas artificiais: Usadas como terapia sintomática para aliviar os sintomas de irritação da superfície.
Uso contínuo de lentes de contato gelatinosas terapêuticas (SCL): Pode suprimir a recorrência de lesões gelatinosas elevadas e prolongar o intervalo cirúrgico.
O uso contínuo de SCL terapêuticas é considerado como terapia conservadora e adjuvante 6). Maeno et al. em 2020, em um estudo observacional prospectivo em pacientes com GDLD, mostraram que o uso de SCL terapêuticas suprime significativamente a recorrência de lesões gelatinosas elevadas de cor cinza a amarela 7). Também é recomendado para prevenção de recorrência pós-operatória.
Ceratectomia superficial terapêutica com laser excimer: Primeira escolha para opacidades superficiais.
Indicações: Lesões gelatinosas elevadas superficiais iniciais a moderadas. Combinado com curetagem manual. Resultados de longo prazo 8,9).
Transplante de Córnea
Transplante superficial, lamelar anterior profundo (DALK) e penetrante total (PKP): Indicado para casos avançados.
Taxa de recorrência: A recorrência após transplante penetrante total é alta, 97% em 4 anos. No DALK, há vantagem de preservação do endotélio.
Transplante de Limbo Corneano
Transplante de células-tronco limbares e formação de epitélio corneano: Usado em conjunto com transplante de córnea.
Objetivo: Cobrir a superfície ocular com epitélio corneano derivado do enxerto e prevenir a reinvasão do epitélio do hospedeiro 10,11).
Resultados de longo prazo da PTK foram relatados no Japão. Ōura et al. mostraram os resultados de longo prazo da PTK em casos de GDLD e relataram sua utilidade em prolongar o tempo até a recorrência 8). Hieda et al., em um estudo multicêntrico japonês, analisaram detalhadamente o tempo de recorrência e os resultados clínicos após PTK9).
A combinação de transplante de células-tronco limbares (LSCT) é uma abordagem originária do Japão reconhecida globalmente. Shimazaki et al. em 2002 relataram a eficácia do transplante de córnea com LSCT para GDLD, mostrando que pode prolongar o tempo até a recorrência ao prevenir a reinvasão de células epiteliais do hospedeiro 10). Posteriormente, Movahedan et al. também relataram abordagem semelhante 11).
Para cobrir a superfície ocular com epitélio corneano derivado do enxerto, o epitélio corneano do hospedeiro é removido e então é realizado o transplante límbico. No pós-operatório, o uso contínuo de lentes de contato terapêuticas é mantido, retardando também a recorrência.
Nos últimos anos, o uso de córnea artificial (Boston tipo I Kpro) também está sendo considerado. Teoricamente, pode evitar a redeposição de amiloide porque não passa pelo epitélio corneano do hospedeiro, mas há risco de complicações pós-operatórias como infecção e membrana retroprotética.
QA recorrência ocorre mesmo após o transplante de córnea?
A
Na GDLD, a taxa de recorrência após o transplante de córnea é extremamente alta. Foi relatado que cerca de 97% recorrem dentro de 4 anos após o transplante penetrante de córnea (PKP). A principal causa é a substituição das células epiteliais do receptor pelo epitélio do enxerto. Como medida originada no Japão, o retardo da recorrência é alcançado pela combinação de transplante de células-tronco límbicas da córnea10) e uso contínuo de lentes de contato terapêuticas 7). No manejo de longo prazo, o objetivo é manter a função visual e prolongar os intervalos cirúrgicos, assumindo a recorrência.
6. Fisiopatologia e Mecanismo Detalhado de Ocorrência
O gene TACSTD2, gene causador da GDLD, é um gene de éxon único localizado no cromossomo 1p32. Foi identificado como gene causador em 1999 por Tsujikawa et al. através de análise de ligação em famílias japonesas 4). A proteína TACSTD2 desempenha um papel essencial na manutenção da função de barreira do epitélio corneano.
Quando ocorrem mutações de perda de função no gene TACSTD2, a localização intracelular normal das proteínas de junção oclusiva Claudina 1 e Claudina 7 é prejudicada. Nakatsukasa et al. mostraram, através da análise de células epiteliais da córnea cultivadas e famílias japonesas, que a perda de função do TACSTD2 leva à perda da localização das Claudinas na junção apicolateral e à diminuição da função de barreira epitelial 5). Além disso, em 2011, relataram novas mutações no TACSTD2 em 3 famílias e sua localização intracelular anormal 12).
Devido à diminuição da função de barreira epitelial, proteínas como a lactoferrina das lágrimas penetram na córnea. A lactoferrina penetrada forma fibrilas amiloides e se deposita sob o epitélio corneano. Os depósitos amiloides contêm lactoferrina, mas esta doença não é devida a uma anormalidade no gene da lactoferrina.
Histologicamente, a opacidade leitosa subepitelial apresenta coloração laranja-avermelhada com vermelho Congo e exibe birrefringência verde-maçã sob microscopia de polarização. Na microscopia eletrônica, as junções oclusivas do epitélio são substituídas por espaços eletrolucentes. Os depósitos também penetram nas lamelas corneanas, causando degeneração das fibras de colágeno e proteoglicanos.
Mais de 20 mutações foram relatadas no gene TACSTD212). No Japão, a mutação Q118X (mutação sem sentido, funcional nula) é uma mutação fundadora que representa mais de 80% dos cromossomos causadores da doença2). A doença geralmente se manifesta em homozigotos, mas também ocorre em heterozigotos compostos devido a casamentos entre famílias diferentes. Curiosamente, mesmo em homozigotos Q118X idênticos, os quatro subtipos (amora, em faixa, kumquat e opacidade parenquimatosa) são observados misturados3).
A amiloidose corneana é classificada como primária ou secundária, sistêmica ou local. A GDLD é classificada como amiloidose local primária juntamente com a distrofia corneana em grade. A degeneração amiloide local secundária ocorre devido a triquíase, ceratocone, trauma ou uso prolongado de lentes de contato, e deve ser diferenciada diagnosticamente.
A GDLD é uma doença rara, e havia escassez de médicos com experiência clínica em cada instituição, além da falta de métodos padronizados de diagnóstico e tratamento. O Grupo de Pesquisa de Levantamento Epidemiológico de Doenças Raras e Intratáveis da Córnea do Ministério da Saúde, Trabalho e Bem-Estar, e o grupo de Pesquisa de Desenvolvimento e Disseminação de Diretrizes Clínicas para Doenças Intratáveis do Segmento Anterior estabeleceram critérios diagnósticos e classificação de gravidade2). Em 2019, a “Distrofia Corneana Gotosa Gelatinosa” foi reconhecida como uma doença rara especificada, e atualmente estão sendo elaboradas diretrizes clínicas baseadas no Minds2).
A longo prazo, a recorrência e os múltiplos tratamentos são problemáticos. A combinação de lentes de contato terapêuticas e transplante límbico pode retardar a recorrência e prolongar os intervalos cirúrgicos, melhorando o prognóstico ao longo da vida6, 7, 10).
Relatos de Casos Atípicos e Recorrência Unilateral
Maeno et al. relataram um caso clínico atípico mostrando depósito amiloide recorrente em apenas um olho, demonstrando a variabilidade fenotípica da GDLD13). O teste genético do TACSTD2 desempenha um papel crucial no diagnóstico desses casos atípicos2).
Na pesquisa básica, espera-se a elucidação detalhada da dinâmica das moléculas de junção estreita a jusante de TACSTD2 e o desenvolvimento de terapias direcionadas à estabilização da Claudina. Clinicamente, a expansão das indicações de abordagens regenerativas, como córnea artificial, transplante de folha de epitélio corneano e epitélio corneano derivado de células iPS, está sendo considerada.
Ide T, Nishida K, Maeda N, Tsujikawa M, Yamamoto S, Watanabe H, et al. A spectrum of clinical manifestations of gelatinous drop-like corneal dystrophy in japan. American journal of ophthalmology. 2004;137(6):1081-4. doi:10.1016/j.ajo.2004.01.048. PMID:15183793.
Tsujikawa M, Kurahashi H, Tanaka T, Nishida K, Shimomura Y, Tano Y, et al. Identification of the gene responsible for gelatinous drop-like corneal dystrophy. Nature genetics. 1999;21(4):420-3. doi:10.1038/7759. PMID:10192395.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Tsujikawa M, et al. Tumor-associated calcium signal transducer 2 is required for the proper subcellular localization of claudin 1 and 7: implications in the pathogenesis of gelatinous drop-like corneal dystrophy. Am J Pathol. 2010;177(3):1344-1355. PMID: 20651236. PMCID: PMC2928967. doi:10.2353/ajpath.2010.100149.
Kawasaki S, Kinoshita S. Clinical and basic aspects of gelatinous drop-like corneal dystrophy. Dev Ophthalmol. 2011;48:97-115. PMID: 21540633. doi:10.1159/000324079.
Maeno S, Soma T, Tsujikawa M, Shigeta R, Kawasaki R, Oie Y, et al. Efficacy of therapeutic soft contact lens in the management of gelatinous drop-like corneal dystrophy. The British journal of ophthalmology. 2020;104(2):241-246. doi:10.1136/bjophthalmol-2018-313809. PMID:31023713.
Hieda O, Kawasaki S, Yamamura K, Nakatsukasa M, Kinoshita S, Sotozono C. Clinical outcomes and time to recurrence of phototherapeutic keratectomy in Japan. Medicine. 2019;98(27):e16216. doi:10.1097/MD.0000000000016216. PMID:31277131; PMCID:PMC6635226.
Shimazaki J, Shimmura S, Tsubota K. Limbal stem cell transplantation for the treatment of subepithelial amyloidosis of the cornea (gelatinous drop-like dystrophy). Cornea. 2002;21(2):177-80. doi:10.1097/00003226-200203000-00010. PMID:11862090.
Nakatsukasa M, Kawasaki S, Yamasaki K, Fukuoka H, Matsuda A, Nishida K, et al. Two novel mutations of TACSTD2 found in three Japanese gelatinous drop-like corneal dystrophy families with their aberrant subcellular localization. Molecular vision. 2011;17:965-70. PMID:21541270; PMCID:PMC3084224.
Maeno S, Soma T, Nishida K. A Case of Clinically Atypical Gelatinous Drop-like Corneal Dystrophy With Unilateral Recurrent Amyloid Depositions. Cornea. 2022;41(11):1447-1450. doi:10.1097/ICO.0000000000003070. PMID:36219213.
Copie o texto do artigo e cole no assistente de IA de sua preferência.
Artigo copiado para a área de transferência
Abra um assistente de IA abaixo e cole o texto copiado na conversa.