A distrofia corneana granular (GCD) é uma doença corneana hereditária caracterizada por depósitos granulares no estroma corneano. Na segunda edição da Classificação Internacional de Distrofias Corneanas (IC3D 2015), é classificada como distrofia epitelial-estromal relacionada ao TGFBI 2).
É causada por mutações pontuais no gene TGFBI (cromossomo 5q31) e segue herança autossômica dominante. Dependendo da mutação, classifica-se em dois tipos:
Classificação
Mutação principal
Sinônimo/nome antigo
Depósito principal
GCD1
Arg555Trp (R555W)
Granular clássica, Groenouw tipo 1
Apenas hialina
GCD2
Arg124His (R124H)
Distrofia corneana de Avellino
Hialina + amiloide
A GCD2 foi relatada em 1988 como um subtipo independente da GCD1, e foi chamada de distrofia corneana de Avellino porque a primeira família era originária da região de Avellino, na Itália 1). Posteriormente, em 1997, o gene causador TGFBI foi identificado e a mutação R124H foi identificada 3). A partir da 2ª edição da classificação IC3D (2015), foi oficialmente denominada distrofia corneana granular tipo 2 (GCD2), e “Avellino” é tratado como um nome histórico, listado em conjunto 2).
Internacionalmente, as distrofias corneanas granulares são classificadas no grupo das distrofias epiteliais-estromais. Este grupo inclui seis doenças relacionadas ao TGFBI: granular (GCD1, GCD2), lattice (LCD1, LCD3A), Reis-Bücklers e Thiel-Behnke, todas causadas por diferentes mutações pontuais no gene TGFBI no mesmo cromossomo 5q31 2,4). Na prática clínica oftalmológica japonesa, essas seis doenças são comumente chamadas coletivamente de “distrofias corneanas relacionadas ao TGFBI”.
A distrofia corneana granular foi relatada pela primeira vez por Groenouw em 1890, e na época era simplesmente chamada de “tipo 1 de Groenouw”. Em 1938, a diferenciação da distrofia lattice foi esclarecida, e por muito tempo foi tratada como uma única doença, “distrofia corneana granular”. Em 1988, um tipo patológico com características tanto granulares quanto lattice foi relatado em uma família da região de Avellino, na Itália, e posteriormente foi separado como GCD2 (tipo Avellino) 1,2). Com a revisão da classificação IC3D em 2015, o sistema de classificação atual foi estabelecido, e a nomenclatura baseada no genótipo tornou-se o padrão internacional 2).
Padrão de herança: Herança autossômica dominante. Apresenta alta penetrância.
Tipo 1: Comum na Europa e América do Norte. Raro no Japão.
Tipo 2: Predominantemente comum no Leste Asiático, como Japão e Coreia. A prevalência na Coreia é de aproximadamente 11,5 por 10.000 pessoas 1).
Proporção entre as distrofias corneanas relacionadas ao TGFBI: GCD2 representa 72-91% na Coreia e Japão, 36% nos EUA e 3% na Polônia 1).
Dados de diagnóstico genético no Japão: Na Universidade de Yamaguchi, durante 21 anos (2000-2021), 234 pacientes com distrofia corneana foram diagnosticados geneticamente, e as quatro principais distrofias corneanas (granular tipo I e II, lattice tipo I e IIIA, gelatinosa em gotas, e macular) representaram cerca de 96% do total. - Características no Leste Asiático: A distrofia corneana granular é predominantemente do tipo 2 (R124H) no Leste Asiático. - Idade de início: Heterozigotos para GCD2 apresentam micro-opacidades observáveis apenas na lâmpada de fenda desde a idade escolar, mas são assintomáticos. A diminuição subjetiva da visão tipicamente aparece entre 40 e 50 anos de idade.
Diferença de sexo: É uma herança autossômica dominante, sem diferença entre sexos.
QO que significa "granular"?
A
Refere-se a uma condição em que múltiplos pequenos aglomerados brancos a branco-acinzentados (depósitos granulares) com bordas bem definidas se formam no estroma superficial da córnea central. Quando observados diretamente com lâmpada de fenda, são descritos como migalhas de pão, flocos de neve ou confeitos. Os depósitos são derivados da proteína TGFBI mutante.
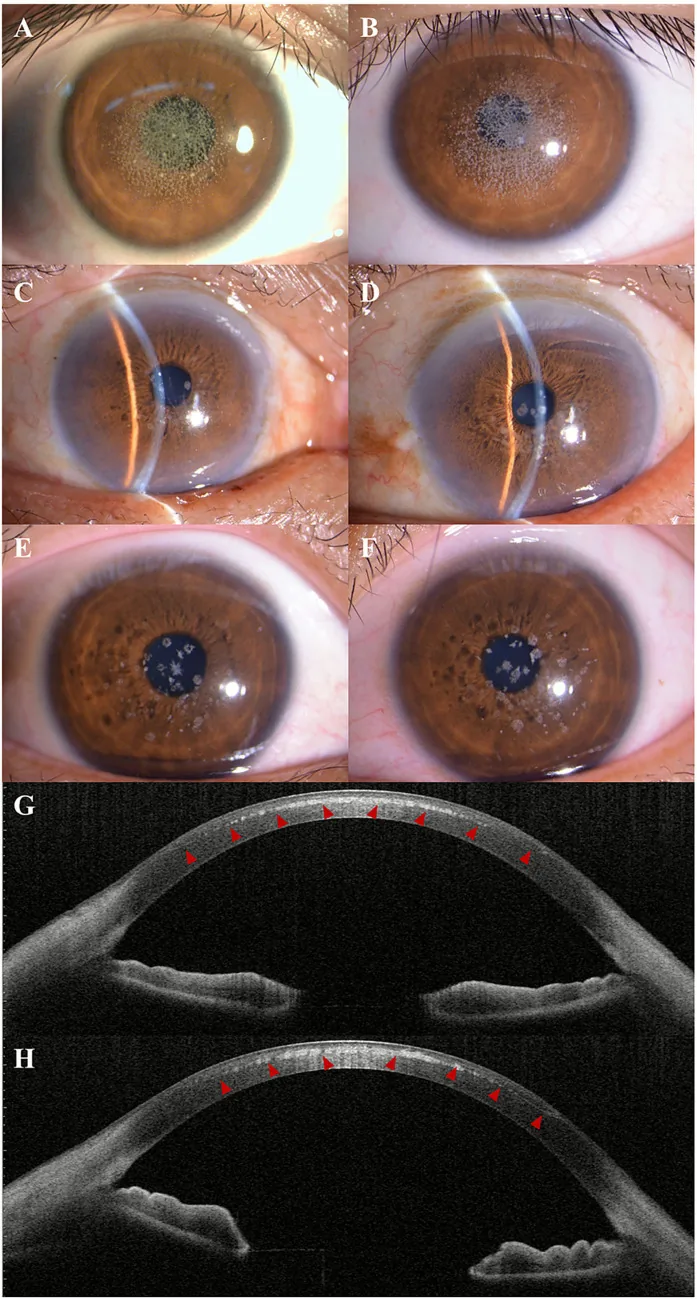
Kuang L, et al. Case Report: Post-LASIK exacerbation of granular corneal dystrophy type 2: a familial case with TGFBI mutation. Front Med (Lausanne). 2025. Figure 1. PMCID: PMC12722859. License: CC BY.
Na fotografia com lâmpada de fenda, opacidades granulares branco-acinzentadas estão dispersas e agrupadas no centro e paracentro da córnea. A AS-OCT mostra depósitos hiper-refletivos no estroma anterior da córnea, demonstrando os achados clínicos da distrofia corneana granular.
Assintomático a leve: Heterozigotos não percebem diminuição da visão nos estágios iniciais a intermediários, e muitos são descobertos incidentalmente em exames de rotina. A maioria começa a relatar diminuição da visão entre 40 e 50 anos.
Glare e fotofobia: Quando as opacidades atingem a área pupilar, os pacientes queixam-se de ofuscamento diurno e diminuição da sensibilidade ao contraste.
Erosão epitelial recorrente: Os depósitos danificam a camada de Bowman e a membrana basal epitelial, causando dor ocular aguda, lacrimejamento e hiperemia durante o sono ou ao acordar.
Diminuição da visão: Quando as áreas transparentes entre os depósitos se tornam opacas, a visão diminui progressivamente3).
Diminuição da sensibilidade ao contraste: Frequentemente, a sensibilidade ao contraste diminui antes da acuidade visual (teste do anel de Landolt).
Tendência à cegueira diurna: Devido ao forte efeito da luz dispersa em ambientes claros, os pacientes queixam-se de ofuscamento ao ar livre ou ao dirigir.
Difícil correção com óculos ou lentes de contato: A dispersão dos depósitos não melhora com a correção refrativa.
Em homozigotos, ocorre diminuição acentuada da visão desde a infância (4-7 anos), necessitando de tratamento por volta dos 10 anos.
Achados clínicos (achados observados pelo médico no exame)
Opacidades granulares: Opacidades granulares brancas a branco-acinzentadas, relativamente pequenas e com bordas nítidas, dispersas na região central da córnea. São descritas como em forma de migalhas de pão ou flocos de neve.
Profundidade: Subepitelial e no estroma superficial da córnea. Não atinge o limbo.
Material depositado: Apenas hialina. Cora-se em vermelho com tricrômio de Masson. Não contém amiloide.
Progressão: Com o envelhecimento, o número de grânulos aumenta e os limites tornam-se indistintos.
GCD2 (R124H)
Opacidades granulares: Iniciam-se como opacidades brancas a branco-acinzentadas, maiores que na GCD1, com bordas nítidas. Os fenótipos são variados: em forma de confete, lineares, estreladas, em bastão, etc.
Tipo misto: Podem ser observadas opacidades lineares finas em forma de rede, semelhantes às da distrofia corneana lattice.
Material depositado: Tanto hialina quanto amiloide. Positivo para tricrômio de Masson e para vermelho Congo, exibindo coloração verde-amarelada à microscopia de polarização.
Progressão: Acima dos 25-30 anos, adicionam-se opacidades brancas densas em forma de bastão e estreladas no estroma médio. A deposição difusa em placa no estroma superficial torna-se mais intensa, sendo boa indicação para PTK3).
Em ambos os tipos, as opacidades localizam-se na região central da córnea, não atingindo a periferia do limbo. Geralmente são bilaterais e com pouca diferença entre os olhos.
Em mutações homozigóticas, o fenótipo é marcadamente diferente.
Homozigoto GCD1: Opacidades reticulares brancas, quase sem espaços, na mesma profundidade, do subepitélio corneano ao estroma superficial. Com a progressão, a íris e a câmara anterior tornam-se invisíveis.
Homozigoto GCD2: Opacidades brancas circulares sólidas em toda a córnea, exceto na periferia extrema. É tão grave que a brancura é perceptível a olho nu, apenas a transparência do limbo é preservada3). Casos homozigotos são distrofias corneanas refratárias que recorrem em curto prazo (1-2 anos) mesmo após PTK ou transplante de córnea.
QQual é a diferença no curso entre homozigotos e heterozigotos?
A
Os homozigotos manifestam-se na infância (4-7 anos) e progridem rapidamente. Opacidades brancas preenchem toda a córnea sem espaços, e PTK ou transplante de córnea são necessários por volta dos 10 anos. Mesmo após a cirurgia, a recorrência ocorre em 1-2 anos, com um curso refratário. Os heterozigotos progridem lentamente e geralmente mantêm boa visão até os 40-50 anos.
A GCD é causada por mutações pontuais no gene TGFBI (cromossomo 5q31). O gene TGFBI codifica a proteína da matriz extracelular TGFBIp (ceratoepitelina). A TGFBIp mutante tem sensibilidade reduzida à degradação proteolítica e acumula-se como depósitos insolúveis anormais no estroma corneano1,5,7).
O grupo de distrofias corneanas associadas ao TGFBI inclui2,4):
Distrofia corneana granular tipo 1 (R555W)
Distrofia corneana granular tipo 2 (R124H, antiga Avellino)
História familiar: É uma herança autossômica dominante, e 50% dos filhos de uma pessoa afetada têm risco de desenvolver a doença.
Homozigose: Indivíduos homozigotos para a mesma mutação apresentam um fenótipo mais grave.
Cirurgia corneana: A GCD2 pode progredir rapidamente após trauma corneano. A opacidade piora significativamente especialmente após cirurgia refrativa a laser 1,8,9).
Raça: A GCD2 é mais comum no Leste Asiático (Coreia, Japão). Suspeita-se do envolvimento de um efeito fundador genético 3).
Fatores ambientais não esclarecidos: A influência direta da exposição aos raios UV ou do diabetes ainda não foi estabelecida.
A GCD é uma doença autossômica dominante com alta penetrância. Quando um caso índice é diagnosticado, 50% dos parentes de primeiro grau (pais, irmãos, filhos) podem ter a mesma mutação. A identificação precoce de portadores assintomáticos na família permite evitar futuras cirurgias refrativas e planejar consultas regulares para monitoramento da progressão 1,5). Especialmente em jovens que desejam realizar LASIK, recomenda-se fortemente uma anamnese cuidadosa da história familiar e, se necessário, teste genético.
O diagnóstico clínico baseia-se na observação de opacidades granulares bem delimitadas no estroma anterior ao biomicroscópio de lâmpada de fenda e em uma história familiar positiva. Suspeita-se de distrofia corneana quando há opacidades (depósitos) corneanas bilaterais sem hiperemia ou edema corneano 3).
No diagnóstico diferencial das distrofias corneanas, primeiro determina-se se os depósitos são “bem delimitados” ou “difusos” 3). Se forem depósitos granulares bem delimitados, diferencia-se GCD1 (pequenos) de GCD2 (grandes) pelo tamanho dos depósitos. Na GCD2, a esclerose difusa revela opacidades em placas difusas entre os depósitos granulares, e essas opacidades em placas são uma boa indicação para PTK3).
Biomicroscopia de lâmpada de fenda: Observa-se diretamente opacidades granulares brancas bem delimitadas. Também são utilizadas a esclerose difusa, a retroiluminação e a transiluminação.
Microscopia confocal: Revela opacidades irregulares, hiper-refletivas, semelhantes a migalhas de pão entre o epitélio e a camada de Bowman.
Microscopia ultrassônica de biomicroscopia (UBM): Detecta grânulos hiper-refletivos no estroma superficial.
Análise da topografia corneana: Fornece informações adicionais sobre a densidade da opacidade.
Teste genético: A análise do gene TGFBI é útil para o diagnóstico definitivo. No Japão, desde abril de 2020, está coberto pelo seguro como teste genético para distrofias corneanas 3).
Fotografia do segmento anterior: Para acompanhamento de longo prazo, é importante tirar fotografias de alta qualidade do segmento anterior na primeira consulta e em cada consulta de acompanhamento, registrando-as no prontuário.
No exame com lâmpada de fenda realizado por profissional experiente, as seguintes técnicas de observação são combinadas para aumentar a sensibilidade 3).
Iluminação direta: Avalia o tamanho e a densidade dos depósitos granulares com bordas bem definidas.
Método de dispersão escleral: Enfatiza as opacidades difusas em placas entre os depósitos. Particularmente útil na GCD2.
Retroiluminação: Utiliza a luz que atravessa a área pupilar para detectar microdepósitos.
GCD1: Depósitos hialinos que se coram em vermelho com tricrômico de Masson. Não contêm amiloide. Na microscopia eletrônica, depósitos em forma de bastonete ou trapézio.
GCD2: Depósitos de hialina (positiva para tricrômico de Masson) e amiloide (positiva para vermelho Congo, verde-amarelado ao microscópio de polarização). Na microscopia eletrônica, observam-se depósitos eletrodensos em forma de bastonete e fibrilas amiloides 1,7).
Distrofia corneana em treliça tipo 1 (LCD1): mutação TGFBI R124C. Opacidades lineares e em treliça devido à deposição de amiloide no estroma. Frequentemente associada a erosões epiteliais recorrentes3)
Distrofia corneana macular (MCD): mutação no gene CHST6. Herança autossômica recessiva. Opacificação difusa de toda a córnea
Distrofia corneana de Reis-Bücklers: mutação TGFBI R124L. Opacidades em forma de mapa na camada de Bowman
Distrofia corneana em pontos (FCD): mutação PIP5K3. Pequenas manchas brancas em todo o estroma, geralmente assintomática
QO teste genético é coberto pelo seguro?
A
Desde abril de 2020, o teste genético para distrofia corneana é coberto pelo seguro. No entanto, requer certificação da instituição, portanto, as instalações capazes de realizar o teste são limitadas. Quando os achados clínicos são suspeitos ou quando se considera cirurgia refrativa como LASIK, o diagnóstico definitivo por teste genético é desejável.
Nos estágios iniciais, sem diminuição da acuidade visual ou erosões epiteliais recorrentes, o tratamento não é necessário. A intervenção cirúrgica é considerada quando a diminuição da visão atinge a área pupilar.
Lágrimas artificiais: colírio de hialuronato de sódio 0,1% ou 0,3%, 4 a 6 vezes ao dia, para reduzir o ressecamento e a irritação
Lentes de contato gelatinosas terapêuticas: para erosões epiteliais recorrentes, protegem a superfície ocular e promovem a cicatrização. Uso contínuo durante o dia, com troca regular
Colírios e pomadas antibióticas: para prevenção de infecção secundária durante erosões epiteliais, usar colírio de levofloxacino 0,5% 3 a 4 vezes ao dia e pomada de ofloxacino ao deitar
Solução salina hipertônica (colírio/pomada oftálmica de cloreto de sódio a 5%): pode ser usada como tratamento adjuvante para reduzir o edema epitelial
Homozigoto GCD2: recidiva cerca de 18 meses após a primeira PTK, e após a segunda, terceira ou mais, recidiva em cerca de 3 meses1)
Heterozigoto GCD2: a recidiva após PTK é relativamente lenta, com média de 38,4 meses1)
Uso concomitante de mitomicina C (MMC): o uso de MMC durante PTK não é recomendado. Isso porque a MMC induz apoptose dos ceratócitos do estroma corneano, reduzindo as células responsáveis pela reabsorção e degradação de TGFBIp, podendo acelerar a recidiva1)
Casos de piora após LASIK: a PTK é possível, mas a eficácia é maior após a remoção do flap de LASIK1,8)
Cuidados pós-operatórios: após PTK, usar colírio antibiótico (levofloxacino 0,5%) e colírio de corticosteroide (fluorometolona 0,1%) 4 vezes ao dia até a cicatrização epitelial, depois reduzir a dose. A cicatrização epitelial geralmente leva de 3 a 5 dias
DALK (Transplante de Córnea Lamelar Profundo) na prática
DALK é uma técnica que preserva o endotélio, utilizando a técnica da grande bolha (big bubble) para dissecar e remover o estroma até logo acima da membrana de Descemet, e suturar o estroma do doador. Como não há risco de rejeição endotelial, o prognóstico a longo prazo é considerado melhor que o PK3). No estudo de Kitazawa et al., a acuidade visual 5 anos após DALK para distrofias corneanas associadas ao TGFBI (incluindo granular e lattice) foi geralmente boa, com alta taxa de sobrevivência do enxerto10). No Japão, é realizável como tratamento coberto pelo seguro saúde.
A escolha do tratamento cirúrgico para GCD é considerada na seguinte ordem1,6).
Depósitos limitados ao estroma anterior (subepitelial a cerca de 150 μm) → PTK
Recidiva após múltiplas PTKs ou opacidade profunda (150 a 400 μm) → DALK
Opacidade em toda a espessura, comprometimento endotelial associado ou casos em que DALK não é possível → PK
Casos refratários com recidivas repetidas após qualquer técnica (especialmente homozigotos GCD2) → considerar abordagens de pesquisa, como terapia genética
A GCD é contraindicação para LASIK, LASEK, PRK e SMILE. Após a cirurgia, a opacidade corneana pode piorar rapidamente, levando a perda grave da visão 1,8,9). Após LASIK, formam-se múltiplos depósitos granulares pequenos entre o flap e o leito estromal. O LASIK resulta em piora mais grave e pior acuidade visual final em comparação com PRK 1,8). Relatos de casos da Coreia e do Japão descrevem muitos pacientes assintomáticos antes da cirurgia que desenvolveram opacidade corneana significativa meses a anos após LASIK, necessitando de PTK ou transplante de córnea8,9).
QO que acontece se a GCD for descoberta após ter feito LASIK?
A
Em casos de GCD após LASIK, formam-se rapidamente depósitos granulares entre o flap e o leito estromal. As opções de tratamento incluem PTK após remoção do flap do LASIK, DALK e PK, a serem consideradas nessa ordem. A consulta precoce com um oftalmologista é importante.
O gene TGFBI codifica a proteína TGFBIp (queratoepitelina, 68 kDa) da matriz extracelular. A TGFBIp está envolvida na adesão, migração e proliferação celular, e é expressa no estroma corneano normal 1,5,7). Mutações no gene TGFBI reduzem a suscetibilidade da TGFBIp mutante à degradação proteolítica, levando ao acúmulo como depósitos insolúveis no estroma corneano5,7).
A GCD2 é quase exclusivamente causada pela mutação Arg124His (R124H) 1,5). Na GCD2, tanto hialina quanto amiloide se depositam.
Defeito na autofagia: Na GCD2, foi relatado defeito na autofagia, reduzindo a degradação da TGFBIp e promovendo seu acúmulo 1,5)
Disfunção mitocondrial: A TGFBIp mutante pode afetar diretamente os fibroblastos corneanos, levando à disfunção mitocondrial 1)
Efeito da neovascularização corneana: Em áreas com neovascularização corneana, os depósitos tendem a diminuir ou ser reabsorvidos. Isso apoia o mecanismo de concentração dos depósitos na região central da córnea, que não possui suprimento vascular1)
Após LASIK, ocorre deposição rápida de TGFBIp entre o flap e o leito estromal. Acredita-se que a manipulação cirúrgica na região central da córnea promova o acúmulo de TGFBIp mutante1,8). Como a incisão corneana na cirurgia de catarata (próxima ao limbo) não agrava a condição, supõe-se que a distância do limbo vascularizado esteja relacionada1). Observações patológicas de Awwad et al. sugerem que os depósitos formados após LASIK se acumulam juntamente com a ativação de queratócitos associada à resposta de cicatrização na interface flap-leito estromal8).
Os depósitos da GCD1 são observados como material eosinofílico homogêneo ao microscópio óptico e coram-se em vermelho com tricrômio de Masson. Em nível de microscopia eletrônica, são identificados como estruturas eletrodensas em forma de bastão ou trapézio, com 100 a 500 nm de diâmetro6).
Na GCD2, além dos depósitos hialinos, observam-se fibrilas amiloides (8 a 10 nm de diâmetro). As fibrilas amiloides coram-se em laranja-avermelhado com vermelho Congo e exibem birrefringência verde-maçã sob microscopia de luz polarizada6). Essa dupla característica de coloração é útil para o diagnóstico patológico definitivo da GCD2.
De acordo com a análise proteômica de Poulsen et al., na córnea de pacientes com GCD2, a TGFBIp mutante R124H é menos suscetível à clivagem por proteases em comparação com a TGFBIp normal, resultando no acúmulo seletivo de fragmentos específicos da extremidade C-terminal6). Sugere-se que essa resistência à clivagem possa ser a base para a formação de fibrilas hialinas e amiloides.
O cloreto de lítio demonstrou reduzir a produção da proteína TGFBI. A terapia combinada de melatonina e rapamicina pode inibir a expressão da proteína TGFBI e, ao mesmo tempo, ativar a autofagia para promover a degradação da TGFBIp mutante1,5).
O silenciamento da expressão do TGFBI mutante usando small interfering RNA (siRNA) ou short hairpin siRNA (shRNA) está sendo estudado em nível pré-clínico. A edição genômica com CRISPR/Cas9 também é uma candidata, mas o efeito off-target não intencional em alelos normais ou outros genes é um desafio1,5).
Eletrólise corneana (corneal electrolysis): O uso experimental em casos de recorrência após transplante de córnea foi relatado. Os resultados a longo prazo são desconhecidos.
Apoio diagnóstico por aprendizado de máquina: O desenvolvimento de um modelo de IA para identificar automaticamente a GCD a partir de fotografias do segmento anterior foi relatado.
Terapia com chaperonas: A pesquisa básica sobre chaperonas químicas (como o ácido 4-fenilbutírico) que auxiliam no dobramento correto da TGFBIp mutante está em andamento3).
Folha de epitélio corneano derivada de células iPS: O transplante de folhas de epitélio corneano produzidas a partir de células iPS do paciente está sendo estudado como uma opção futura.
No Japão, discussões continuam sobre a construção de um registro nacional de distrofias corneanas relacionadas ao TGFBI, lideradas pela Sociedade Japonesa de Oftalmologia e pela Sociedade Japonesa de Córnea. Desde a cobertura do seguro para testes genéticos em 2020, o número de casos diagnosticados geneticamente aumentou, e dados de acompanhamento de longo prazo sobre padrões de mutação e fenótipos específicos dos japoneses estão sendo acumulados3,11). Espera-se o desenvolvimento de um sistema clínico baseado em evidências, desde a triagem de portadores até a avaliação de indicações para cirurgia refrativa.
Chang MS, Jun I, Kim EK. Mini-Review: Clinical Features and Management of Granular Corneal Dystrophy Type 2. Korean journal of ophthalmology : KJO. 2023;37(4):340-347. doi:10.3341/kjo.2023.0032. PMID:37336511; PMCID:PMC10427907.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Lisch W, Weiss JS. Clinical and genetic update of corneal dystrophies. Experimental eye research. 2019;186:107715. doi:10.1016/j.exer.2019.107715. PMID:31301286.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern®. Ophthalmology. 2024;131(4):P1-P79.
Poulsen ET, Dyrlund TF, Runager K, et al. Proteomics of granular corneal dystrophy type 2 reveals altered TGFBIp proteolytic processing. J Proteome Res. 2014;13(10):4659-4667.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Awwad ST, Di Pascuale MA, Hogan RN, Forstot SL, McCulley JP, Cavanagh HD. Avellino corneal dystrophy worsening after laser in situ keratomileusis: further clinicopathologic observations and proposed pathogenesis. American journal of ophthalmology. 2008;145(4):656-61. doi:10.1016/j.ajo.2007.12.008. PMID:18243154; PMCID:PMC2661203.
Jun RM, Tchah H, Kim TI, et al. Avellino corneal dystrophy after LASIK. Ophthalmology. 2004;111(3):463-468.
Kitazawa K, Kawasaki S, Shinomiya K, et al. Safety of anterior lamellar keratoplasty for recurrent TGFBI corneal dystrophy. Br J Ophthalmol. 2016;100(4):448-454.