Die granuläre Hornhautdystrophie (GCD) ist eine erbliche Hornhauterkrankung, die durch körnige Ablagerungen im Hornhautstroma gekennzeichnet ist. In der zweiten internationalen Klassifikation der Hornhautdystrophien (IC3D 2015) wird sie zu den epithelial-stromalen TGFBI-assoziierten Dystrophien gezählt 2).
Ursache sind Punktmutationen im TGFBI-Gen (Chromosom 5q31), die autosomal-dominant vererbt werden. Je nach Mutation werden die folgenden zwei Typen unterschieden.
Klassifikation
Hauptmutation
Synonym / frühere Bezeichnung
Hauptablagerungen
GCD1
Arg555Trp (R555W)
Klassisch granulär / Groenouw Typ 1
Nur Hyalin
GCD2
Arg124His (R124H)
Avellino-Korneadystrophie
Hyalin + Amyloid
GCD2 wurde 1988 als Subtyp von GCD1 beschrieben, und da die erste Familie aus der italienischen Region Avellino stammte, wurde sie als Avellino-Korneadystrophie bezeichnet 1). 1997 wurde das verantwortliche Gen TGFBI identifiziert und die R124H-Mutation nachgewiesen 3). Seit der zweiten Auflage der IC3D-Klassifikation (2015) wird sie offiziell als granuläre Korneadystrophie Typ 2 (GCD2) bezeichnet, und „Avellino“ wird als historischer Name parallel geführt 2).
Die granuläre Korneadystrophie wird international zu den epithelial-stromalen Dystrophien gezählt. Diese Gruppe umfasst sechs TGFBI-assoziierte Erkrankungen: granuläre (GCD1, GCD2), gittrige (LCD1, LCD3A), Reis-Bücklers und Thiel-Behnke, die alle durch unterschiedliche Punktmutationen im TGFBI-Gen auf Chromosom 5q31 verursacht werden 2,4). In der japanischen ophthalmologischen Praxis werden diese sechs Erkrankungen üblicherweise als „TGFBI-assoziierte Korneadystrophien“ zusammengefasst.
Die granuläre Korneadystrophie wurde erstmals 1890 von Groenouw beschrieben und damals einfach als „Groenouw Typ 1“ bezeichnet. 1938 wurde die Abgrenzung zur gittrigen Dystrophie geklärt, und lange Zeit wurde sie als einheitliche Erkrankung „granuläre Korneadystrophie“ behandelt. 1988 wurde in einer Familie aus der italienischen Region Avellino ein Krankheitstyp mit Merkmalen sowohl der granulären als auch der gittrigen Dystrophie beschrieben, der später als GCD2 (Avellino-Typ) abgetrennt wurde 1,2). Mit der Überarbeitung der IC3D-Klassifikation 2015 wurde das aktuelle Klassifikationssystem etabliert, und die genotypbasierte Nomenklatur wurde zum internationalen Standard 2).
Vererbungsmodus: autosomal-dominant mit hoher Penetranz
Typ 1: Häufig in Europa und Nordamerika, selten in Japan
Typ 2: Überwiegend in Ostasien (Japan, Korea). Die Prävalenz in Korea beträgt etwa 11,5 pro 10.000 Einwohner 1)
Anteil an TGFBI-assoziierten Korneadystrophien: GCD2 macht in Korea und Japan 72–91 %, in den USA 36 % und in Polen 3 % aus 1)
Japanische genetische Diagnosedaten: An der Yamaguchi-Universität wurden in 21 Jahren (2000–2021) 234 Patienten mit Hornhautdystrophie genetisch diagnostiziert. Die vier häufigsten Hornhautdystrophien (granuläre Typ I und II, gittrige Typ I und IIIA, gelatinöse tropfenförmige und fleckige) machten etwa 96 % aller Fälle aus. – Besonderheiten in Ostasien: Die granuläre Hornhautdystrophie ist in Ostasien überwiegend vom Typ 2 (R124H). – Erkrankungsalter: Heterozygote Träger der GCD2 zeigen bereits im Schulalter nur unter der Spaltlampe sichtbare Mikrotrübungen, jedoch keine subjektiven Symptome. Eine subjektive Sehverschlechterung tritt typischerweise im Alter von 40–50 Jahren auf.
Geschlechterverteilung: autosomal-dominante Vererbung, daher kein Geschlechterunterschied.
QWas bedeutet „granulär“?
A
Es bezeichnet den Zustand, bei dem sich in den oberflächlichen Schichten des zentralen Hornhautstromas zahlreiche scharf begrenzte, weiße bis grauweiße kleine Klumpen (granuläre Ablagerungen) bilden. Unter direkter Spaltlampenmikroskopie werden sie als bröselig, schneeflockenartig oder kandiszuckerartig beschrieben. Die Ablagerungen bestehen aus mutiertem TGFBI-Protein.
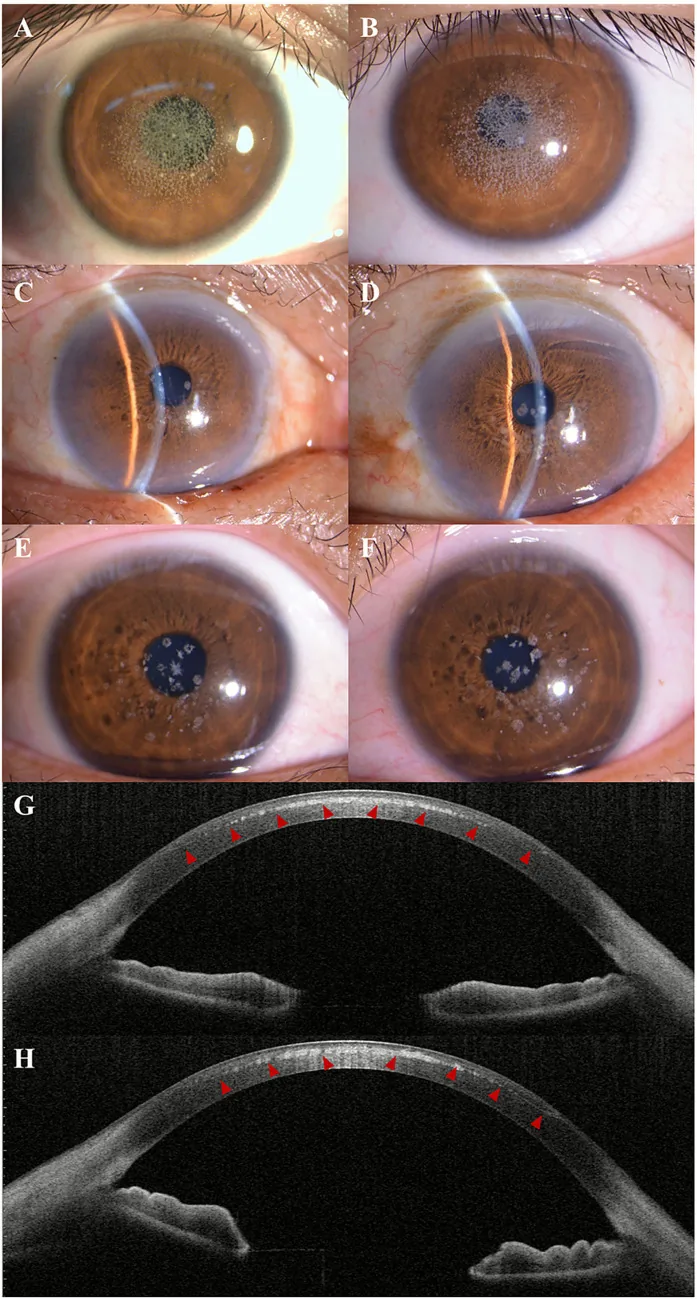
Kuang L, et al. Case Report: Post-LASIK exacerbation of granular corneal dystrophy type 2: a familial case with TGFBI mutation. Front Med (Lausanne). 2025. Figure 1. PMCID: PMC12722859. License: CC BY.
Im Spaltlampenfoto sind von der Hornhautmitte bis zur parazentralen Region verstreute und gruppierte grauweiße granuläre Trübungen zu sehen. Im AS-OCT zeigen sich hyperreflektive Ablagerungen im vorderen Hornhautstroma, was den klinischen Befund einer granulären Hornhautdystrophie darstellt.
Asymptomatisch bis leicht: Heterozygote Träger bemerken im frühen bis mittleren Stadium keine Sehverschlechterung; nicht selten wird die Erkrankung zufällig bei Vorsorgeuntersuchungen entdeckt. Meist wird erst im Alter von 40–50 Jahren eine Sehverschlechterung beklagt.
Blendung und Photophobie: Wenn die Trübungen die Pupillenregion betreffen, klagen die Patienten über Blendung am Tag und verminderten Kontrast.
Rezidivierende Hornhauterosionen: Die Ablagerungen schädigen die Bowman-Schicht und die Basalmembran des Epithels, was zu stechenden Augenschmerzen, Tränenfluss und Rötung während des Schlafs oder beim Aufwachen führt.
Sehverschlechterung: Wenn die transparenten Bereiche zwischen den Ablagerungen undurchsichtig werden, nimmt die Sehkraft fortschreitend ab3).
Verminderte Kontrastempfindlichkeit: Häufig sinkt die Kontrastempfindlichkeit bereits vor der Sehschärfe (gemessen mit Landolt-Ringen).
Neigung zur Nachtblindheit: Da bei Helligkeit Streulicht stark stört, klagen Patienten über Blendung im Freien oder beim Autofahren.
Schwierige Korrektur mit Brille oder Kontaktlinsen: Die Streuung durch die Ablagerungen lässt sich durch Brechungskorrektur nicht verbessern.
Bei Homozygoten kommt es bereits im Kindesalter (4–7 Jahre) zu einer deutlichen Sehverschlechterung, und im Alter von etwa 10 Jahren ist eine Behandlung erforderlich.
Klinische Befunde (Befunde, die der Arzt bei der Untersuchung feststellt)
Körnige Trübungen: In der zentralen Hornhaut verstreute, scharf begrenzte, relativ kleine, weiße bis grauweiße körnige Trübungen. Sie werden als bröselig oder schneeflockenartig beschrieben.
Tiefe: Subepithelial und im oberflächlichen Stroma der Hornhaut. Der Limbus ist nicht betroffen.
Ablagerungssubstanz: Nur Hyalin. Färbt sich mit Masson-Trichrom rot. Enthält kein Amyloid.
Progression: Mit zunehmendem Alter nimmt die Anzahl der Körner zu, und die Grenzen werden unscharf.
GCD2 (R124H)
Körnige Trübungen: Beginnt mit größeren, weißen bis grauweißen, scharf begrenzten Trübungen als bei GCD1. Die Phänotypen sind vielfältig: kandiszuckerartig, linear, sternförmig, keulenförmig usw.
Mischtyp: Es können auch netzartige, feine lineare Trübungen auftreten, wie sie bei der gittrigen Hornhautdystrophie vorkommen.
Ablagerungssubstanz: Sowohl Hyalin als auch Amyloid. Masson-Trichrom-positiv und Kongorot-positiv, unter dem Polarisationsmikroskop gelbgrün.
Progression: Ab einem Alter von 25–30 Jahren kommen im mittleren Stroma dichte, weiße, stäbchen- und sternförmige Trübungen hinzu. Im oberflächlichen Stroma nehmen diffuse flächige Ablagerungen zu, was eine gute Indikation für PTK darstellt 3).
Bei beiden Typen liegen die Trübungen in der zentralen Hornhaut und reichen nicht bis zum Limbus. In der Regel sind beide Augen betroffen, mit geringen Seitenunterschieden.
Bei homozygoten Mutationen unterscheidet sich der Phänotyp erheblich.
GCD1-Homozygot: Weiße, netzartige Trübungen in nahezu lückenloser Anordnung in derselben Tiefe unter dem Epithel bis zum oberflächlichen Stroma. Im fortgeschrittenen Stadium werden Iris und Vorderkammer unsichtbar.
GCD2-Homozygot: Dichte, runde, weiße Trübungen, die die gesamte Hornhaut mit Ausnahme des äußersten Randes lückenlos bedecken. So schwerwiegend, dass die Trübung bereits mit bloßem Auge erkennbar ist; nur die Limbusregion bleibt klar 3). Homozygote Fälle sind eine therapieresistente Hornhautdystrophie, die auch nach PTK oder Hornhauttransplantation innerhalb von 1–2 Jahren rezidiviert.
QWie unterscheidet sich der Verlauf bei Homozygoten und Heterozygoten?
A
Homozygote erkranken im Kindesalter (4–7 Jahre) mit schnellem Fortschreiten. Es kommt zu lückenlosen weißen Trübungen der gesamten Hornhaut, sodass im Alter von etwa 10 Jahren eine PTK oder Hornhauttransplantation erforderlich wird. Auch postoperativ rezidiviert die Erkrankung innerhalb von 1–2 Jahren und verläuft therapieresistent. Heterozygote hingegen schreiten langsam voran und können in der Regel bis zum 40.–50. Lebensjahr eine gute Sehkraft aufrechterhalten.
GCD wird durch Punktmutationen im TGFBI-Gen (Chromosom 5q31) verursacht. Das TGFBI-Gen kodiert für das extrazelluläre Matrixprotein TGFBIp (Keratoepithelin). Mutiertes TGFBIp ist weniger anfällig für den Proteinabbau und reichert sich als abnorme unlösliche Ablagerungen im Hornhautstroma an 1,5,7).
Zur TGFBI-assoziierten Hornhautdystrophie-Gruppe gehören 2,4):
Granuläre Hornhautdystrophie Typ 1 (R555W)
Granuläre Hornhautdystrophie Typ 2 (R124H, früher Avellino)
Gitterförmige Hornhautdystrophie Typ 1 (R124C)
Gitterförmige Hornhautdystrophie Typ 3A (R501T oder L527R)
Familienanamnese: autosomal-dominante Vererbung, 50 % der Kinder eines Betroffenen haben ein Risiko, die Erkrankung zu entwickeln
Homozygotie: Homozygote für dieselbe Mutation zeigen einen schwereren Phänotyp
Hornhautchirurgie: GCD2 kann nach Hornhautverletzungen schnell fortschreiten. Besonders nach Laser-Refraktionschirurgie verschlechtert sich die Trübung deutlich 1,8,9)
Ethnische Zugehörigkeit: GCD2 ist in Ostasien (Korea, Japan) häufig. Es wird ein Gründereffekt vermutet 3)
Umweltfaktoren ungeklärt: Ein direkter Einfluss von UV-Exposition oder Diabetes ist derzeit nicht belegt
GCD ist eine autosomal-dominante Erkrankung mit hoher Penetranz. Wenn bei einem Indexpatienten die Diagnose gesichert ist, besteht bei 50 % der erstgradigen Verwandten (Eltern, Geschwister, Kinder) die Möglichkeit, dieselbe Mutation zu tragen. Die frühzeitige Identifizierung asymptomatischer Anlageträger in der Familie ermöglicht es, zukünftige refraktive Chirurgie zu vermeiden und regelmäßige Nachsorgeuntersuchungen zur Verlaufsbeobachtung zu planen 1,5). Besonders bei jungen Menschen, die eine LASIK wünschen, werden eine sorgfältige Erhebung der Familienanamnese und gegebenenfalls ein Gentest dringend empfohlen.
Die klinische Diagnose basiert auf der Beobachtung scharf begrenzter granulärer Trübungen im vorderen Stroma mittels Spaltlampenmikroskopie und einer positiven Familienanamnese. Bei beidseitigen Hornhauttrübungen (Ablagerungen) ohne Rötung und Hornhautödem sollte eine Hornhautdystrophie in Betracht gezogen werden 3).
Bei der Differenzialdiagnose von Hornhautdystrophien wird zunächst beurteilt, ob die Ablagerungen „scharf begrenzt“ oder „diffus“ sind 3). Bei scharf begrenzten granulären Ablagerungen wird anhand der Größe zwischen GCD1 (klein) und GCD2 (groß) unterschieden. Bei GCD2 können mittels Sklerastreulicht diffuse flächige Trübungen zwischen den granulären Ablagerungen festgestellt werden; diese flächigen Trübungen sind eine gute Indikation für eine PTK3).
Spaltlampenmikroskopie: Direkte Beobachtung scharf begrenzter weißer granulärer Trübungen. Auch Sklerastreulicht, retrograde Beleuchtung und Durchleuchtung werden genutzt.
Vorderabschnitts-OCT (AS-OCT): Zeigt hochreflektive Trübungen im vorderen Stroma. Nützlich für die Planung der Ablationstiefe bei PTK.
Konfokale Mikroskopie: Unregelmäßige, hochreflektive krümelige Trübungen zwischen Epithel und Bowman-Schicht.
Hornhauttopographie: Liefert zusätzliche Informationen zur Dichte der Trübung.
Gentest: TGFBI-Genanalyse ist für die definitive Diagnose nützlich. In Japan seit April 2020 als Hornhautdystrophie-Gentest von der Krankenkasse erstattet 3).
Vorderabschnittsfotografie: Für die Langzeitbeobachtung ist es wichtig, bei der Erstuntersuchung und bei jeder Nachuntersuchung qualitativ hochwertige Vorderabschnittsfotos zu machen und in der Krankenakte zu dokumentieren.
GCD1: Hyalinablagerungen, die sich mit Masson-Trichrom rot färben. Kein Amyloid. Elektronenmikroskopisch stäbchen- oder trapezförmige Ablagerungen.
GCD2: Sowohl Hyalin (Masson-Trichrom-positiv) als auch Amyloid (Kongorot-positiv, im Polarisationsmikroskop gelbgrün) lagern sich ab. Elektronenmikroskopisch stäbchenförmige elektronendichte Ablagerungen und Amyloidfibrillen 1,7).
Gitterförmige Hornhautdystrophie Typ 1 (LCD1): TGFBI R124C-Mutation. Lineare und gitterförmige Trübungen durch Amyloidablagerungen im Stroma. Häufig begleitet von rezidivierenden Hornhauterosionen 3)
Fleckförmige Hornhautdystrophie (MCD): CHST6-Genmutation. Autosomal-rezessiv. Diffuse Trübung der gesamten Hornhaut
Reis-Bücklers-Hornhautdystrophie: TGFBI R124L-Mutation. Landkartenartige Trübungen der Bowman-Schicht
Fleckchen-Hornhautdystrophie (FCD): PIP5K3-Mutation. Kleine weiße Flecken im gesamten Stroma, meist asymptomatisch
QWird der Gentest von der Krankenkasse übernommen?
A
Seit April 2020 ist der Gentest für Hornhautdystrophien als Kassenleistung aufgeführt. Allerdings ist eine Einrichtungszertifizierung erforderlich, sodass nur begrenzte Einrichtungen den Test durchführen können. Bei verdächtigen klinischen Befunden oder wenn eine refraktive Korrektur wie LASIK in Betracht gezogen wird, ist eine definitive Diagnose durch Gentest wünschenswert.
In frühen Stadien ohne Sehverschlechterung oder rezidivierende Hornhauterosionen ist keine Behandlung erforderlich. Wenn die Sehverschlechterung den Pupillarbereich erreicht, wird ein chirurgischer Eingriff in Betracht gezogen.
Künstliche Tränen: Natriumhyaluronat 0,1 % oder 0,3 % Augentropfen 4- bis 6-mal täglich zur Linderung von Trockenheit und Reizung
Therapeutische weiche Kontaktlinsen: Bei rezidivierenden Erosionen schützen sie die Augenoberfläche und fördern die Heilung. In der Regel ganztägiges Tragen, regelmäßiger Austausch erforderlich
Antibiotische Augentropfen/-salbe: Zur Sekundärinfektionsprophylaxe bei Erosionen: Levofloxacin 0,5 % Augentropfen 3- bis 4-mal täglich, vor dem Schlafengehen Ofloxacin-Augensalbe
Hypertonische Kochsalzlösung (5% Natriumchlorid-Augentropfen/-salbe): Kann als unterstützende Behandlung zur Reduktion des Epithelödems eingesetzt werden
Die Behandlung wird je nach Tiefe der Ablagerungen gewählt. Das ultimative Ziel ist es, eine Hornhauttransplantation so lange wie möglich hinauszuzögern1,6).
PTK (Therapeutische Keratektomie)
Indikation: Trübungen im vorderen Stroma. Wird als Erstlinientherapie empfohlen1).
Vorteile: Wiederholbar. Kein Risiko einer Transplantatabstoßung. Diffuse subepitheliale Trübungen sind eine gute Indikation für PTK3).
Technik: Mit dem Excimer-Laser werden pro Sitzung etwa 50 μm Stroma abgetragen. Die Ablationstiefe ist durch die Hornhautdicke begrenzt1,8).
Grenzen: Pro Abtragung kommt es zu einer Hyperopisierung von etwa 1,5 dpt. Die Anzahl der Behandlungen ist begrenzt.
DALK/ALK (Tiefe lamelläre Keratoplastik)
Indikation: Tiefe Stromatrübungen, wenn PTK aufgrund der Hornhautdicke nicht durchführbar ist.
Vorteile: DALK wird bevorzugt, da das Hornhautendothel normal ist. Kein Risiko einer endothelialen Abstoßungsreaktion3).
Rezidiv: Rezidive treten an der Wirts-Transplantat-Grenze oder im oberflächlichen Stroma des Transplantats auf3).
PK (Perforierende Keratoplastik)
Indikation: Ultima Ratio bei wiederholten Rezidiven mit tiefen Stromaablagerungen.
Mediane Zeit bis zum Rezidiv: PK hat mit 13,7 Jahren die längste Dauer. PTK, ALK und DALK liegen bei 2,7–3,7 Jahren1).
Endgültige korrigierte Sehschärfe: Bei allen Operationsmethoden vergleichbar (20/25–20/30) 1).
GCD2-Homozygoten: Rezidiv etwa 18 Monate nach der ersten PTK, nach der zweiten, dritten und weiteren Behandlungen etwa 3 Monate 1)
GCD2-Heterozygoten: Rezidiv nach PTK im Durchschnitt nach 38,4 Monaten, relativ langsam 1)
Mitomycin C (MMC)-Kombination: Die Anwendung von MMC bei PTK wird nicht empfohlen, da MMC die Apoptose von Keratozyten im Hornhautstroma induziert und die Zellen reduziert, die für die Resorption und den Abbau von TGFBIp verantwortlich sind, was das Rezidiv beschleunigen kann 1)
Verschlechterung nach LASIK: PTK ist möglich, aber die Wirkung ist nach Entfernung des LASIK-Flaps besser 1,8)
Postoperative Behandlung: Nach PTK werden antibiotische Augentropfen (Levofloxacin 0,5 %) und Kortikosteroid-Augentropfen (Fluorometholon 0,1 %) bis zur Epithelheilung 4-mal täglich, dann reduziert, angewendet. Die Epithelheilung dauert in der Regel 3–5 Tage.
DALK (tiefe lamelläre Keratoplastik) in der Praxis
Die DALK ist ein Verfahren, das das Endothel erhält. Mit der Big-Bubble-Technik wird das Stroma bis unmittelbar über der Descemet-Membran abgelöst und entfernt, und das Spenderstroma wird eingenäht. Da kein Risiko einer Endothelabstoßung besteht, wird die Langzeitprognose als besser als bei der PK angesehen 3). In einer Studie von Kitazawa et al. war die 5-Jahres-Sehschärfe nach DALK bei TGFBI-assoziierter Hornhautdystrophie (einschließlich granulärer und gittriger Formen) im Allgemeinen gut, und die Transplantatüberlebensrate war hoch 10). In Japan ist die Behandlung im Rahmen der Krankenversicherung möglich.
Flussdiagramm zur Auswahl des Operationsverfahrens
Die Auswahl der chirurgischen Behandlung bei GCD wird in der folgenden Reihenfolge geprüft 1,6).
Ablagerungen auf das vordere Stroma (subepithelial bis etwa 150 μm) beschränkt → PTK
Rezidiv nach mehrfacher PTK oder Trübungen in der Tiefe (150–400 μm) → DALK
Trübungen in allen Schichten, begleitende Endothelschädigung, Fälle, in denen DALK nicht durchgeführt werden kann → PK
Therapieresistente Fälle mit wiederholten Rezidiven nach jedem Verfahren (insbesondere GCD2-Homozygoten) → Prüfung eines experimentellen Ansatzes wie Gentherapie
Kontraindikationen für die laserbasierte refraktive Chirurgie
GCD ist eine Kontraindikation für LASIK, LASEK, PRK und SMILE. Nach dem Eingriff kann es zu einer raschen Verschlechterung der Hornhauttrübung und schwerer Sehverschlechterung kommen 1,8,9). Nach LASIK bilden sich zahlreiche kleine körnige Ablagerungen zwischen dem Flap und dem Stromabett. LASIK führt im Vergleich zu PRK zu einer schwereren Verschlechterung und schlechteren endgültigen Sehschärfe1,8). Fallberichte aus Korea und Japan beschreiben zahlreiche Fälle, bei denen zuvor asymptomatische Patienten Monate bis Jahre nach LASIK eine deutliche Hornhauttrübung entwickelten und eine PTK oder Hornhauttransplantation benötigten 8,9).
QWas passiert, wenn GCD nach einer LASIK festgestellt wird?
A
Bei Fällen von GCD nach LASIK bilden sich schnell körnige Ablagerungen zwischen dem Flap und dem Stromabett. Zu den Behandlungsoptionen gehören PTK nach Entfernung des LASIK-Flaps, DALK und PK, die in dieser Reihenfolge in Betracht gezogen werden. Eine frühzeitige Konsultation eines Augenarztes ist wichtig.
6. Pathophysiologie und detaillierter Entstehungsmechanismus
Das TGFBI-Gen kodiert für das extrazelluläre Matrixprotein TGFBIp (Keratoepithelin, 68 kDa). TGFBIp ist an Zelladhäsion, -migration und -proliferation beteiligt und wird auch im normalen Hornhautstroma exprimiert 1,5,7). Mutationen im TGFBI-Gen führen zu einer verminderten Empfindlichkeit des mutierten TGFBIp gegenüber Proteolyse, was zu einer Akkumulation als unlösliche Ablagerungen im Hornhautstroma führt 5,7).
GCD1 wird durch die Arg555Trp (R555W)-Mutation verursacht. Das mutierte TGFBIp lagert sich als Hyalin in den oberflächlichen Schichten des Hornhautstromas ab. Amyloid ist nicht enthalten 3).
GCD2 ist fast ausschließlich auf die Arg124His (R124H)-Mutation beschränkt 1,5). Bei GCD2 lagern sich sowohl Hyalin als auch Amyloid ab.
Autophagiestörung: Bei GCD2 wurde eine Störung der Autophagie berichtet, die zu einer verminderten Degradation von TGFBIp und damit zu verstärkter Akkumulation führt 1,5)
Mitochondriale Dysfunktion: Es wird vermutet, dass das mutierte TGFBIp selbst Hornhautfibroblasten beeinflusst und eine mitochondriale Dysfunktion verursachen kann 1)
Einfluss von Hornhautneovaskularisation: In Bereichen mit Hornhautneovaskularisation neigen Ablagerungen dazu, abzunehmen oder resorbiert zu werden. Dies unterstützt den Mechanismus, dass sich Ablagerungen im avaskulären zentralen Hornhautbereich konzentrieren 1).
Nach LASIK lagert sich TGFBIp schnell zwischen dem Flap und dem Stromabett ab. Dies wird darauf zurückgeführt, dass der chirurgische Eingriff im zentralen Hornhautbereich die Akkumulation von mutiertem TGFBIp fördert 1,8). Da sich die Hornhautinzision bei Kataraktoperationen (in der Nähe des Limbus) nicht verschlechtert, wird angenommen, dass der Abstand zum vaskularisierten Limbus eine Rolle spielt 1). Die pathologischen Beobachtungen von Awwad et al. deuten darauf hin, dass die nach LASIK gebildeten Ablagerungen mit der Aktivierung von Keratozyten im Rahmen der Wundheilungsreaktion an der Grenzfläche zwischen Flap und Stromabett sowie der Ansammlung von TGFBIp zusammenhängen 8).
Die Ablagerungen bei GCD1 erscheinen im Lichtmikroskop als homogene eosinophile Substanz und färben sich mit Masson-Trichrom rot. Auf elektronenmikroskopischer Ebene werden sie als stäbchen- oder trapezförmige elektronendichte Strukturen mit einem Durchmesser von 100–500 nm erkannt 6).
Bei GCD2 finden sich neben Hyalinablagerungen auch Amyloidfibrillen (Durchmesser 8–10 nm). Amyloidfibrillen färben sich mit Kongorot orangerot und zeigen unter dem Polarisationsmikroskop eine apfelgrüne Doppelbrechung 6). Die doppelte Färbeeigenschaft ist für die pathologische Diagnose von GCD2 nützlich.
Laut Proteomanalysen von Poulsen et al. wird mutiertes R124H-TGFBIp in der Hornhaut von GCD2-Patienten im Vergleich zu normalem TGFBIp weniger leicht durch Proteasen gespalten, und bestimmte C-terminale Fragmente reichern sich selektiv an 6). Es wird vermutet, dass diese Spaltungsresistenz die Grundlage für die Bildung von Hyalin- und Amyloidfibrillen bilden könnte.
Lithiumchlorid wurde berichtet, die TGFBI-Proteinproduktion zu reduzieren. Die Kombinationstherapie mit Melatonin und Rapamycin könnte die TGFBI-Protein expression hemmen und gleichzeitig die Autophagie aktivieren, um den Abbau von mutiertem TGFBIp zu fördern 1,5).
Das Silencing der mutierten TGFBI-Expression mittels small interfering RNA (siRNA) oder short hairpin siRNA (shRNA) wird präklinisch untersucht. Die Genom-Editierung mit CRISPR/Cas9 ist ebenfalls ein Kandidat, jedoch sind unbeabsichtigte Off-Target-Effekte auf normale Allele oder andere Gene eine Herausforderung 1,5).
Hornhautelektrolyse (corneal electrolysis): Ein experimenteller Einsatz bei Rezidiven nach Hornhauttransplantation wurde berichtet. Langzeitergebnisse sind unbekannt.
Diagnoseunterstützung durch maschinelles Lernen: Die Entwicklung eines KI-Modells zur automatischen Erkennung von GCD aus Vorderabschnittsfotos wurde berichtet.
Chaperon-Therapie: Grundlagenforschung zu chemischen Chaperonen (z. B. 4-Phenylbuttersäure), die die korrekte Faltung von mutiertem TGFBIp unterstützen, ist im Gange3).
iPS-Zell-abgeleitete Hornhautepithelschicht: Die Transplantation von aus patienteneigenen iPS-Zellen hergestellten Hornhautepithelschichten wird als zukünftige Option erforscht.
Im Inland werden unter der Leitung der Japanischen Ophthalmologischen Gesellschaft und der Japanischen Hornhautgesellschaft weiterhin Diskussionen über den Aufbau eines nationalen Registers für TGFBI-assoziierte Hornhautdystrophien geführt. Seit der Aufnahme von Gentests in die Krankenversicherung im Jahr 2020 hat die Zahl der genetisch diagnostizierten Fälle zugenommen, und es werden zunehmend Langzeit-Follow-up-Daten zu für Japaner spezifischen Mutationsmustern und Phänotypen gesammelt3,11). In Zukunft wird ein evidenzbasiertes klinisches System erwartet, das von der Träger-Screening bis zur Beurteilung der Indikation für refraktive Chirurgie reicht.
Bei Patienten mit GCD sind die folgenden Lebensstilhinweise wichtig.
UV-Schutz: Tragen Sie beim Ausgehen eine Sonnenbrille oder Brille mit UV-Schutz, um die UV-Belastung der Hornhaut zu reduzieren.
Vermeidung von Augenverletzungen: Prellungen oder Fremdkörper können die Hornhaut schädigen und rezidivierende Erosiones auslösen. Tragen Sie bei Sport oder Arbeit eine Schutzbrille.
Regelmäßige Kontrolluntersuchungen: Auch Heterozygote sollten einmal jährlich eine Spaltlampenuntersuchung und Sehtest erhalten.
Familienuntersuchung: Empfehlen Sie erstgradigen Verwandten ein familiäres Screening.
Kontaktlinsen: Weiche Linsen sind grundsätzlich möglich, aber die Tragedauer sollte kurz gehalten und ein regelmäßiger Austausch strikt eingehalten werden.
Vermeidung refraktiver Chirurgie: LASIK-ähnliche Operationen sind absolut kontraindiziert. Wählen Sie eine Korrektur mit Brille oder Kontaktlinsen.
Chang MS, Jun I, Kim EK. Mini-Review: Clinical Features and Management of Granular Corneal Dystrophy Type 2. Korean journal of ophthalmology : KJO. 2023;37(4):340-347. doi:10.3341/kjo.2023.0032. PMID:37336511; PMCID:PMC10427907.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Lisch W, Weiss JS. Clinical and genetic update of corneal dystrophies. Experimental eye research. 2019;186:107715. doi:10.1016/j.exer.2019.107715. PMID:31301286.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern®. Ophthalmology. 2024;131(4):P1-P79.
Poulsen ET, Dyrlund TF, Runager K, et al. Proteomics of granular corneal dystrophy type 2 reveals altered TGFBIp proteolytic processing. J Proteome Res. 2014;13(10):4659-4667.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Awwad ST, Di Pascuale MA, Hogan RN, Forstot SL, McCulley JP, Cavanagh HD. Avellino corneal dystrophy worsening after laser in situ keratomileusis: further clinicopathologic observations and proposed pathogenesis. American journal of ophthalmology. 2008;145(4):656-61. doi:10.1016/j.ajo.2007.12.008. PMID:18243154; PMCID:PMC2661203.
Jun RM, Tchah H, Kim TI, et al. Avellino corneal dystrophy after LASIK. Ophthalmology. 2004;111(3):463-468.
Kitazawa K, Kawasaki S, Shinomiya K, et al. Safety of anterior lamellar keratoplasty for recurrent TGFBI corneal dystrophy. Br J Ophthalmol. 2016;100(4):448-454.