這是一種由TGFBI基因突變引起的體染色體顯性遺傳 角膜失養症 ,導致角膜基質 中透明質和類澱粉蛋白沉積。
分為第1型(R555W突變,Groenouw 1型)和第2型(R124H突變,舊稱Avellino角膜失養症 )。
在日本,第2型壓倒性地多。韓國的盛行率約為每1萬人中有11.5人1) 。
根據山口大學的基因診斷研究,四大角膜失養症 (顆粒狀I/II型、格子狀I/IIIA型、膠滴狀、斑狀)約佔所有病例的96%。- 同合子患者在兒童早期(4-7歲)發病且病情嚴重。異合子患者進展緩慢。
PTK (治療性角膜切除術 )是第一線治療。對於深層混濁,選擇DALK 。LASIK 、PRK、LASEK、SMILE 等雷射屈光 矯正手術為禁忌。 顆粒狀角膜失養症 (GCD )是一種遺傳性角膜 疾病,特徵為角膜基質 中出現顆粒狀沉積物。在國際角膜失養症 分類第二版(IC3D 2015)中,它被歸類為上皮-基質TGFBI相關失養症2) 。
該病由TGFBI基因(染色體5q31)的點突變引起,呈體染色體顯性遺傳 。根據突變不同,分為以下兩型。
分類 主要突變 別名/舊稱 主要沉積物 GCD 1Arg555Trp (R555W) 經典顆粒狀 / Groenouw 第1型 僅透明質 GCD 2Arg124His(R124H) 阿韋利諾角膜失養症 透明蛋白+類澱粉蛋白
GCD 2於1988年被報告為獨立於GCD 1的亞型,因首個家系源自義大利阿韋利諾地區,故稱為阿韋利諾角膜失養症 1) 。隨後在1997年,致病基因TGFBI被確認,並鑑定出R124H突變3) 。自IC3D分類第二版(2015年)起,正式命名為顆粒狀角膜失養症 2型(GCD 2),而「阿韋利諾」作為歷史名稱並列標註2) 。
顆粒狀角膜失養症 在國際上被歸類於上皮-基質失養症群。該群包括六種TGFBI相關疾病:顆粒狀(GCD 1、GCD 2)、格子狀(LCD1、LCD3A)、Reis-Bücklers和Thiel-Behnke,均由5q31上TGFBI基因的不同點突變引起2,4) 。在日本眼科臨床中,這六種疾病統稱為「TGFBI相關角膜失養症 」。
顆粒狀角膜失養症 於1890年由Groenouw首次報告,當時僅稱為「Groenouw 1型」。1938年與格子狀失養症明確區分,長期作為單一疾病「顆粒狀角膜失養症 」處理。1988年,義大利阿韋利諾地區的一個家系報告了兼具顆粒狀和格子狀特徵的病型,後分離為GCD 2(阿韋利諾型)1,2) 。2015年IC3D分類修訂確立了現行分類體系,基於基因型的命名成為國際標準2) 。
遺傳方式 :體染色體顯性遺傳 ,外顯率高1型 :歐美多見,日本少見2型 :日本、韓國等東亞地區占絕對多數。韓國盛行率約為每萬人11.5人1) 在TGFBI相關角膜失養症 中的占比 :GCD 2在韓國和日本占72-91%,美國占36%,波蘭占3%1) 日本基因診斷數據 :山口大學在2000年至2021年的21年間對234例角膜 營養不良患者進行了基因診斷,四大角膜 營養不良(顆粒狀I型、II型,格子狀I型、IIIA型,膠滴狀,斑狀)約佔全部病例的96%。- 東亞特徵 :顆粒狀角膜營養不良 在東亞以2型(R124H)為主。- 發病年齡 :雜合子GCD 2從學齡期起僅在裂隙燈 下可見微小混濁,但無自覺症狀。自覺視力 下降通常出現在40~50歲。性別差異 :體染色體顯性遺傳 ,無性別差異。
Q
「顆粒狀」指的是什麼狀態?
A
指角膜 中央部實質淺層出現許多邊界清晰的白色至灰白色小塊(顆粒狀沉積物)。用裂隙燈 顯微鏡直接觀察時,形容為麵包屑狀、雪花狀或金平糖狀。沉積物來源於突變的TGFBI蛋白。
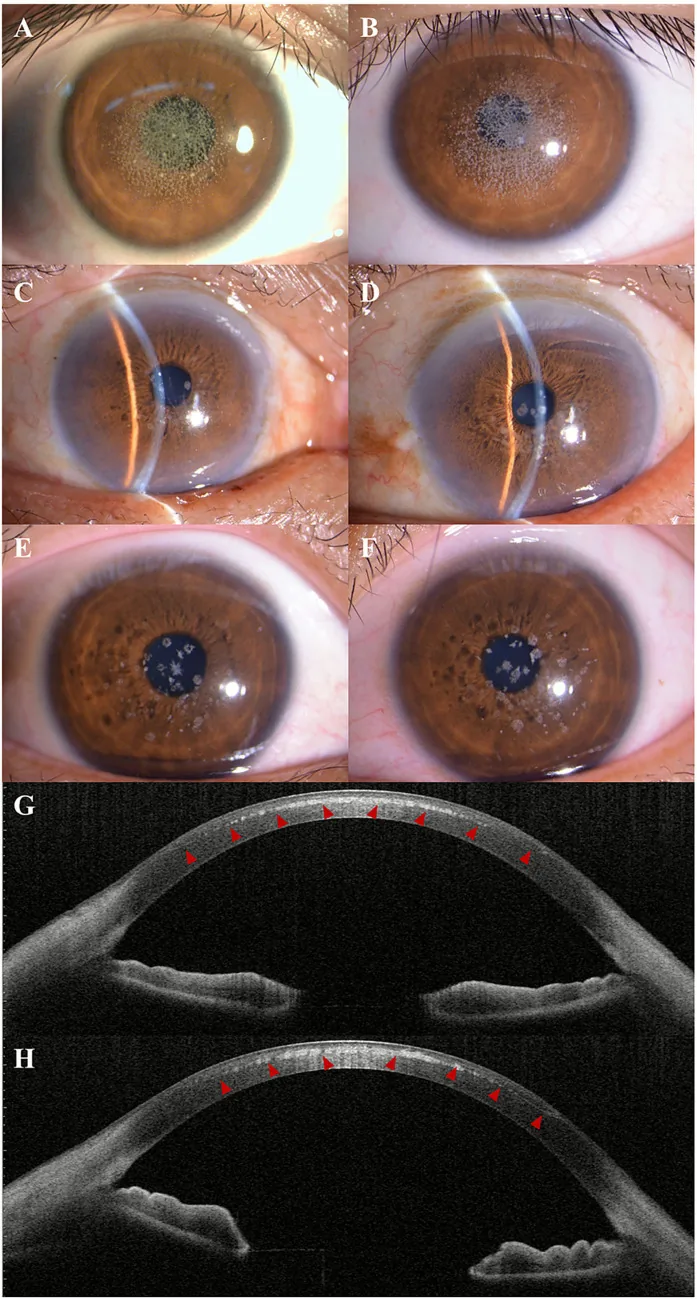
Kuang L, et al. Case Report: Post-LAS
IK exacerbation of granular corneal dystrophy type 2: a familial case with TGFBI mutation. Front Med (Lausanne). 2025. Figure 1. PM
CI D: PMC12722859. License: CC BY.
無症狀至輕度 :雜合子早期至中期常無自覺視力 下降,不少病例在體檢中偶然發現。多數在40~50歲後主訴視力 下降。眩光、畏光 :混濁波及瞳孔 區時,主訴白天眩光和對比敏感度 下降。復發性角膜上皮糜爛 :沉積物損傷鮑曼層和上皮基底膜,睡眠中或起床時出現劇烈眼痛 、流淚、充血 。視力 下降視力 進行性下降3) 。對比敏感度 下降對比敏感度 常先於視力 (蘭多爾特環檢查)下降。晝盲傾向 :明亮環境下散射光影響強烈,因此在戶外或駕駛時主訴眩光。眼鏡或隱形眼鏡難以矯正 :沉積物的散射無法透過屈光 矯正改善。
純合子患者從幼年(4-7歲)開始出現明顯的視力 下降,大約在10歲前後需要治療。
GCD1(R555W)
顆粒狀混濁 :角膜 中央部散在邊界清晰、相對較小的白色至灰白色顆粒狀混濁。描述為麵包屑狀或雪片狀。
深度 :角膜上皮 下及角膜基質 淺層。不累及角膜緣 。
沉積物 :僅透明蛋白。Masson三色染色呈紅色。不含澱粉樣蛋白。
進展 :隨著年齡增長,顆粒數量增加,邊界變得不明確。
GCD2(R124H)
顆粒狀混濁 :發病時混濁比GCD 1更大,呈白色至灰白色,邊界清晰。表型多樣,如金平糖狀、線狀、星狀、棒狀等。
混合型 :有時可見格子狀角膜營養不良 中出現的網狀細線狀混濁。
沉積物 :同時含有透明蛋白和澱粉樣蛋白。Masson三色染色陽性且剛果紅染色陽性,偏振光顯微鏡下呈黃綠色。
進展 :25-30歲以後,基質中層出現棒狀、星狀的濃密白色混濁。淺層瀰漫性片狀沉積增強,是PTK 的良好適應症3) 。
兩種類型的混濁均位於角膜 中央部,不累及角膜緣 周邊部。通常為雙眼性,左右差異較小。
純合子突變的表現型顯著不同。
GCD 1純合子角膜上皮 下至實質淺層同一深度,存在幾乎無間隙的白色網狀混濁。進展後虹膜 與前房 無法觀察。GCD 2純合子角膜 全面出現無間隙的實性圓形白色混濁。嚴重到肉眼可辨識白色,僅角膜緣 透明性得以保留3) 。純合子病例即使進行PTK 或角膜移植 後,也會在1-2年的短期內復發,屬於難治性角膜 營養不良。
Q
純合子與雜合子的病程有何不同?
A
純合子在幼兒期(4-7歲)發病,進展迅速。角膜 全面出現無間隙的白色混濁,10歲左右需要PTK 或角膜移植 。術後1-2年復發,呈難治性病程。雜合子緩慢進展,通常到40-50歲仍能維持良好視力 。
GCD 由TGFBI基因(染色體5q31)的點突變引起。TGFBI基因編碼細胞外基質蛋白TGFBIp(角膜上皮 素)。突變TGFBIp對蛋白水解的敏感性降低,在角膜基質 內作為異常不溶性沉積物積累1,5,7) 。
TGFBI相關角膜 營養不良包括以下類型2,4) :
家族史 :體染色體顯性遺傳 ,患者子女的50%有發病風險。同型合子 :同一突變的同型合子表現更嚴重的表型。角膜 手術GCD 2在角膜 外傷後可快速進展。尤其在雷射屈光 矯正手術後混濁明顯加重1,8,9) 。種族 :GCD 2在東亞(韓國、日本)多見。有研究指出存在遺傳創始者效應3) 。環境因素未明 :紫外線暴露或糖尿病的直接影響目前尚未確定。
GCD 是一種高外顯率的體染色體顯性遺傳 病。若先證者確診,其一級親屬(父母、兄弟姐妹、子女)有50%可能攜帶相同突變。早期識別家族中無症狀攜帶者,有助於避免未來進行屈光 矯正手術,並制定定期追蹤計畫以監測進展1,5) 。尤其對於考慮LASIK 的年輕人,強烈建議詳細詢問家族史並在必要時進行基因檢測。
臨床診斷基於裂隙燈 顯微鏡下觀察到前基質層邊界清晰的顆粒狀混濁,以及陽性家族史。若出現雙側角膜 混濁(沉積物)而無充血 和角膜水腫 ,應懷疑角膜 營養不良3) 。
在角膜 營養不良的鑑別診斷中,首先判斷沉積物是「邊界清晰」還是「瀰漫性」3) 。對於邊界清晰的顆粒狀沉積物,根據沉積物大小區分GCD 1(小)和GCD 2(大)。在GCD 2中,鞏膜 散射法可在顆粒狀沉積物之間觀察到瀰漫性片狀混濁,這種片狀混濁是PTK 的良好適應症3) 。
裂隙燈 顯微鏡鞏膜 散射法、後照法和透照法。前段光學同調斷層掃描 (AS-OCT ) :顯示前基質層的高反射混濁。有助於規劃PTK 切除深度。共軛焦顯微鏡 超音波生物顯微鏡 (UBM )角膜地形圖 基因檢測 :TGFBI基因分析有助於確診。在日本,自2020年4月起,作為角膜 營養不良基因檢測納入健保 3) 。前段照相 :為長期追蹤,初診及每次追蹤時拍攝高品質前段照片並留存於病歷中非常重要。
熟練的裂隙燈 檢查中,結合以下觀察方法可提高敏感度 3) 。
直接照明 :評估邊界清晰的顆粒狀沉積物的大小和密度。鞏膜 散射法GCD 2尤其有用。後部反光法 :利用通過瞳孔 區的光線檢測微小沉積物。透照法 :早期發現上皮下輕微的顆粒狀混濁。
GCD 1GCD 21,7) 。
格子狀角膜營養不良 1型(LCD1)角膜上皮糜爛 3) 斑狀角膜 營養不良(MCD ) :CHS T6基因突變。體染色體隱性遺傳 。角膜 瀰漫性混濁Reis-Bücklers角膜 營養不良 :TGFBI R124L突變。Bowman層 地圖狀混濁Thiel-Behnke角膜 營養不良 :TGFBI R555Q突變。蜂巢狀混濁Schnyder角膜營養不良 角膜 混濁斑點狀角膜營養不良 (FCD)
Q
基因檢測有健保給付嗎?
A
自2020年4月起,角膜 營養不良基因檢測已納入健保給付。但需機構認證,因此可執行檢測的機構有限。當臨床發現可疑或考慮進行LASIK 等屈光 矯正手術時,建議透過基因檢測確診。
在視力 下降或復發性角膜上皮 脫落尚未出現的早期階段,無需治療。當視力 下降累及瞳孔 區時,考慮手術介入。
人工淚液 :使用0.1%或0.3%玻尿酸鈉滴眼液,每日4~6次,減輕乾燥和刺激治療性軟式隱形眼鏡 :針對復發性上皮脫落,保護眼表並促進癒合。通常全天佩戴,需定期更換抗菌滴眼液/眼藥膏 :預防上皮脫落時的繼發感染,使用0.5%左氧氟沙星滴眼液每日3~4次,睡前使用氧氟沙星眼藥膏高張食鹽水(5%氯化鈉眼藥水/眼藥膏) :可作為輔助治療減輕上皮水腫。
根據沉積深度選擇治療方法。最終目標是盡可能延遲角膜移植 1,6) 。
PTK(治療性角膜切除術)
適應症 :前基質混濁。建議作為第一線治療1) 。
優點 :可重複進行。無移植排斥風險。上皮下瀰漫性混濁是PTK 的良好適應症3) 。
操作 :準分子雷射每次切除約50 μm基質。切除深度受角膜 厚度限制1,8) 。
限制 :每次切除導致約1.5 D遠視 。手術次數有限。
DALK/ALK(深層表層角膜移植術)
適應症 :深基質混濁。當PTK 因角膜 厚度限制無法進行時。
優點 :由於角膜內皮 正常,DALK 更受青睞。無內皮型排斥反應風險3) 。
復發 :復發發生在宿主-移植物交界處或移植物淺層基質3) 。
PK(穿透性角膜移植術)
適應症 :反覆復發導致深層基質沉積時的最後手段。
復發中位時間 :PK最長,為13.7年。PTK 、ALK和DALK 為2.7至3.7年1) 。
最終矯正視力 :所有術式效果相當(20/25至20/30)1) 。
GCD 2純合子PTK 後約18個月復發,第二次及以後約3個月復發1) 。GCD 2雜合子PTK 後復發相對緩慢,平均38.4個月1) 。合併使用絲裂黴素C(MMC) :不建議在PTK 時使用MMC。MMC誘導角膜基質 中角膜 細胞的凋亡,減少負責TGFBIp再吸收和降解的細胞,可能加速復發1) 。LASIK 後加重病例 :可行PTK ,但去除LASIK 瓣後效果更佳1,8) 。術後管理 :PTK 後使用抗菌眼藥水(左氧氟沙星0.5%)和皮質類固醇 眼藥水(氟米龍0.1%),每日四次直至上皮癒合,然後減量。上皮癒合通常需要3至5天。
DALK 是一種保留內皮的術式。採用大氣泡技術,將基質剝離至緊貼後彈力層上方,縫合供體基質。由於沒有內皮排斥反應的風險,長期預後優於PK3) 。Kitazawa等人的研究顯示,TGFBI相關角膜 營養不良(包括顆粒狀和格子狀)患者DALK 術後5年視力 普遍良好,植片存活率高10) 。在日本,該手術可在健保下進行。
GCD 的手術治療選擇按以下順序考慮1,6) 。
沉積物侷限於前部基質(上皮下至約150μm)→ PTK
多次PTK 後復發或深層混濁(150–400μm)→ DALK
全層混濁、合併內皮功能障礙或無法行DALK 的病例→ PK
任何術後反覆復發的難治性病例(尤其是GCD 2純合子)→ 考慮基因治療 等研究性方法
GCD 是LASIK 、LASEK、PRK和SMILE 的禁忌症。術後角膜 混濁會迅速惡化,導致嚴重視力 下降1,8,9) 。LASIK 術後,角膜 瓣與基質床之間會形成大量小顆粒狀沉積物。與PRK相比,LASIK 的惡化更嚴重,最終視力 也更差1,8) 。來自韓國和日本的病例報告描述了許多術前無症狀的患者,在LASIK 術後數月至數年內出現明顯的角膜 混濁,需要PTK 或角膜移植 8,9) 。
Q
如果在接受LASIK後發現GCD,會怎麼樣?
A
在LASIK 術後發生GCD 的病例中,角膜 瓣與基質床之間會迅速形成顆粒狀沉積物。治療選擇包括LASIK 角膜 瓣移除後的PTK 、DALK 和PK,按此順序考慮。早期就診眼科專科醫生非常重要。
TGFBI基因編碼細胞外基質蛋白TGFBIp(角膜上皮 素,68 kDa)。TGFBIp參與細胞黏附、遷移和增殖,並在正常角膜基質 中表現1,5,7) 。TGFBI基因發生突變後,突變型TGFBIp對蛋白水解的敏感性降低,導致其作為不溶性沉積物在角膜基質 中蓄積5,7) 。
GCD 1由Arg555Trp(R555W)突變引起。突變型TGFBIp作為透明蛋白沉積在角膜基質 淺層。不含澱粉樣蛋白3) 。
GCD 2幾乎完全由Arg124His(R124H)突變引起1,5) 。在GCD 2中,透明蛋白和澱粉樣蛋白均有沉積。
自噬障礙 :GCD 2中已報導存在自噬障礙,導致TGFBIp降解減少,從而促進蓄積1,5) 粒線體功能障礙 :有研究表明,突變型TGFBIp本身會影響角膜 纖維母細胞,可能導致粒線體功能障礙1) 角膜新生血管 的影響角膜新生血管 的區域,沉積物有減少和再吸收的趨勢。這一發現支持了沉積物集中在無血管的角膜 中央部的機制1)
LASIK 術後,TGFBIp迅速沉積在角膜 瓣和基質床之間。這被認為是由於角膜 中央部的手術操作促進了突變TGFBIp的積累1,8) 。白內障 手術時的角膜 切口(近角膜緣 )不會導致惡化,因此推測與血管化的角膜緣 的距離有關1) 。Awwad等人的病理學觀察表明,LASIK 術後形成的沉積物與角膜 瓣-基質床界面傷口癒合反應相關的角膜 細胞活化以及TGFBIp的聚集有關8) 。
在GCD 1中,沉積物在光學顯微鏡下表現為均質的嗜酸性物質,Masson三色染色呈紅色。在電子顯微鏡水平,它們被識別為直徑100-500nm的棒狀或梯形高電子密度結構6) 。
在GCD 2中,除了透明沉積物外,還觀察到澱粉樣纖維(直徑8-10nm)。澱粉樣纖維經剛果紅染色呈橙紅色,在偏振光顯微鏡下呈現蘋果綠色雙折射6) 。雙重染色特性有助於GCD 2的病理確診。
根據Poulsen等人的蛋白質組學分析,GCD 2患者角膜 中的突變R124H-TGFBIp與正常TGFBIp相比,不易被蛋白水解酶切割,並且特定的C末端片段選擇性積累6) 。這種切割抵抗性被認為是透明和澱粉樣纖維形成的基礎。
針對GCD 2的新療法正在開發中,但均處於研究階段1,5) 。
氯化鋰已被報導可減少TGFBI蛋白的產生。褪黑激素和雷帕黴素的聯合治療可能抑制TGFBI蛋白表達,同時激活自噬以促進突變TGFBIp的降解1,5) 。
使用小干擾RNA(siRNA)或短髮夾RNA(shRNA)沉默突變TGFBI表達的研究正在進行臨床前研究。CRISPR/Cas9基因組編輯技術也是一個候選,但對正常等位基因或其他基因的意外脫靶效劑仍是一個挑戰1,5) 。
角膜 電解術角膜移植術 後復發病例的試驗性使用。長期結果未知。機器學習輔助診斷 :已有報告開發出從前段照片自動識別GCD 的AI模型。分子伴侶療法 :關於化學分子伴侶(如4-苯基丁酸)輔助突變TGFBIp正確折疊的基礎研究正在進行中3) 。iPS細胞來源角膜上皮 片 :使用患者來源iPS細胞製備的角膜上皮 片進行移植治療作為未來選擇正在研究中。
在日本,以日本眼科學會和日本角膜 學會為中心,關於建立TGFBI相關角膜 營養不良國家登記處的討論仍在繼續。自2020年基因檢測納入保險以來,基因診斷病例增加,日本人特有的突變模式和表型的長期追蹤數據正在累積3,11) 。未來,預計將建立從攜帶者篩查到屈光 手術適應症判斷的實證診療體系。
對於GCD 患者,以下生活指導很重要。
紫外線防護 :外出時佩戴具有防紫外線功能的太陽眼鏡或眼鏡,減少角膜 的紫外線暴露。避免眼外傷 :撞擊或異物造成的角膜 損傷可能誘發復發性上皮脫落。運動或工作時使用防護眼鏡。定期檢查 :即使是雜合子也應每年進行一次裂隙燈 檢查和視力檢查 。家族檢查 :建議一級親屬進行家族內篩查。隱形眼鏡 :原則上可以使用軟性隱形眼鏡,但應縮短佩戴時間並嚴格執行定期更換。避免屈光 手術 :LASIK 類手術絕對禁忌。選擇眼鏡或隱形眼鏡矯正。
Chang MS , Jun I, Kim EK. Mini-Review: Clinical Features and Management of Granular Corneal Dystrophy Type 2. Korean journal of ophthalmology : KJO. 2023;37(4):340-347. doi:10.3341/kjo.2023.0032. PMID:37336511; PMCI D:PMC10427907.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Lisch W, Weiss JS. Clinical and genetic update of corneal dystrophies. Experimental eye research. 2019;186:107715. doi:10.1016/j.exer.2019.107715. PMID:31301286.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern®. Ophthalmology. 2024;131(4):P1-P79.
Poulsen ET, Dyrlund TF, Runager K, et al. Proteomics of granular corneal dystrophy type 2 reveals altered TGFBIp proteolytic processing. J Proteome Res. 2014;13(10):4659-4667.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Awwad ST, Di Pascuale MA, Hogan RN, Forstot SL, McCulley JP, Cavanagh HD. Avellino corneal dystrophy worsening after laser in situ keratomileusis: further clinicopathologic observations and proposed pathogenesis. American journal of ophthalmology. 2008;145(4):656-61. doi:10.1016/j.ajo.2007.12.008. PMID:18243154; PMCI D:PMC2661203.
Jun RM, Tchah H, Kim TI, et al. Avellino corneal dystrophy after LASIK . Ophthalmology. 2004;111(3):463-468.
Kitazawa K, Kawasaki S, Shinomiya K, et al. Safety of anterior lamellar keratoplasty for recurrent TGFBI corneal dystrophy. Br J Ophthalmol. 2016;100(4):448-454.
日本眼科学会. 角膜 ジストロフィ遺伝子検査の保険収載について. 日本眼科学会雑誌. 2020;124:厚生労働省告示に基づく2020年4月1日収載.