La dystrophie cornéenne granulaire (GCD) est une maladie cornéenne héréditaire caractérisée par des dépôts granulaires dans le stroma cornéen. Selon la deuxième classification internationale des dystrophies cornéennes (IC3D 2015), elle est classée parmi les dystrophies épithéliales-stromales liées à TGFBI2).
Elle est causée par une mutation ponctuelle du gène TGFBI (chromosome 5q31) et suit un mode de transmission autosomique dominant. Selon la mutation, elle est classée en deux types :
Classification
Mutation principale
Autre nom / Ancien nom
Dépôt principal
GCD1
Arg555Trp (R555W)
Granulaire classique / Groenouw type 1
Hyaline uniquement
GCD2
Arg124His (R124H)
Dystrophie cornéenne d’Avellino
Hyaline + amyloïde
La GCD2 a été rapportée en 1988 comme un sous-type distinct de la GCD1, et a été appelée dystrophie cornéenne d’Avellino car la première famille était originaire de la région d’Avellino en Italie 1). En 1997, le gène responsable TGFBI a été identifié et la mutation R124H a été découverte 3). Depuis la 2e édition de la classification IC3D (2015), elle est officiellement nommée dystrophie cornéenne granuleuse de type 2 (GCD2), et « Avellino » est mentionné comme nom historique 2).
La dystrophie cornéenne granuleuse est classée internationalement dans le groupe des dystrophies épithéliales-stromales. Ce groupe comprend six maladies liées au TGFBI : les formes granuleuses (GCD1, GCD2), les formes réticulaires (LCD1, LCD3A), la dystrophie de Reis-Bücklers et celle de Thiel-Behnke, toutes causées par différentes mutations ponctuelles du gène TGFBI situé sur le chromosome 5q31 2,4). Dans la pratique ophtalmologique japonaise, ces six maladies sont souvent regroupées sous le terme « dystrophies cornéennes liées au TGFBI ».
La dystrophie cornéenne granuleuse a été rapportée pour la première fois par Groenouw en 1890, et était alors simplement appelée « type 1 de Groenouw ». En 1938, la distinction avec la dystrophie réticulaire a été clarifiée, et elle a longtemps été traitée comme une maladie unique sous le nom de « dystrophie cornéenne granuleuse ». En 1988, une forme présentant à la fois des caractéristiques granuleuses et réticulaires a été rapportée dans une famille de la région d’Avellino en Italie, et a ensuite été séparée sous le nom de GCD2 (type Avellino) 1,2). La révision de la classification IC3D en 2015 a établi le système de classification actuel, et la dénomination basée sur le génotype est devenue la norme internationale 2).
Mode de transmission : Autosomique dominant. Pénétrance élevée.
Type 1 : Fréquent en Europe et en Amérique du Nord. Rare au Japon.
Type 2 : Très fréquent au Japon, en Corée et en Asie de l’Est. La prévalence en Corée est d’environ 11,5 personnes pour 10 000 1).
Proportion parmi les dystrophies cornéennes liées au TGFBI : La GCD2 représente 72 à 91 % en Corée et au Japon, 36 % aux États-Unis et 3 % en Pologne 1).
Données de diagnostic génétique au Japon : À l’Université de Yamaguchi, sur 21 ans (2000-2021), 234 patients atteints de dystrophie cornéenne ont été diagnostiqués génétiquement, et les quatre principales dystrophies cornéennes (granulaire type I et II, réticulaire type I et IIIA, gélatineuse en gouttes, maculaire) représentaient environ 96 % du total. - Caractéristiques en Asie de l’Est : La dystrophie cornéenne granulaire est majoritairement de type 2 (R124H) en Asie de l’Est. - Âge d’apparition : Les hétérozygotes pour GCD2 présentent des opacités microscopiques visibles uniquement à la lampe à fente dès l’âge scolaire, mais sans symptômes subjectifs. La baisse d’acuité visuelle subjective apparaît typiquement entre 40 et 50 ans.
Différence de sexe : Il s’agit d’une maladie autosomique dominante, sans différence de sexe.
QQue signifie « granulaire » ?
A
Cela désigne un état où de nombreuses petites masses blanches à blanc-grisâtre (dépôts granulaires) aux limites nettes se forment dans le stroma superficiel central de la cornée. Observées directement au microscope à lampe à fente, elles sont décrites comme des miettes de pain, des flocons de neige ou des bonbons dragéifiés. Les dépôts proviennent de la protéine TGFBI mutée.
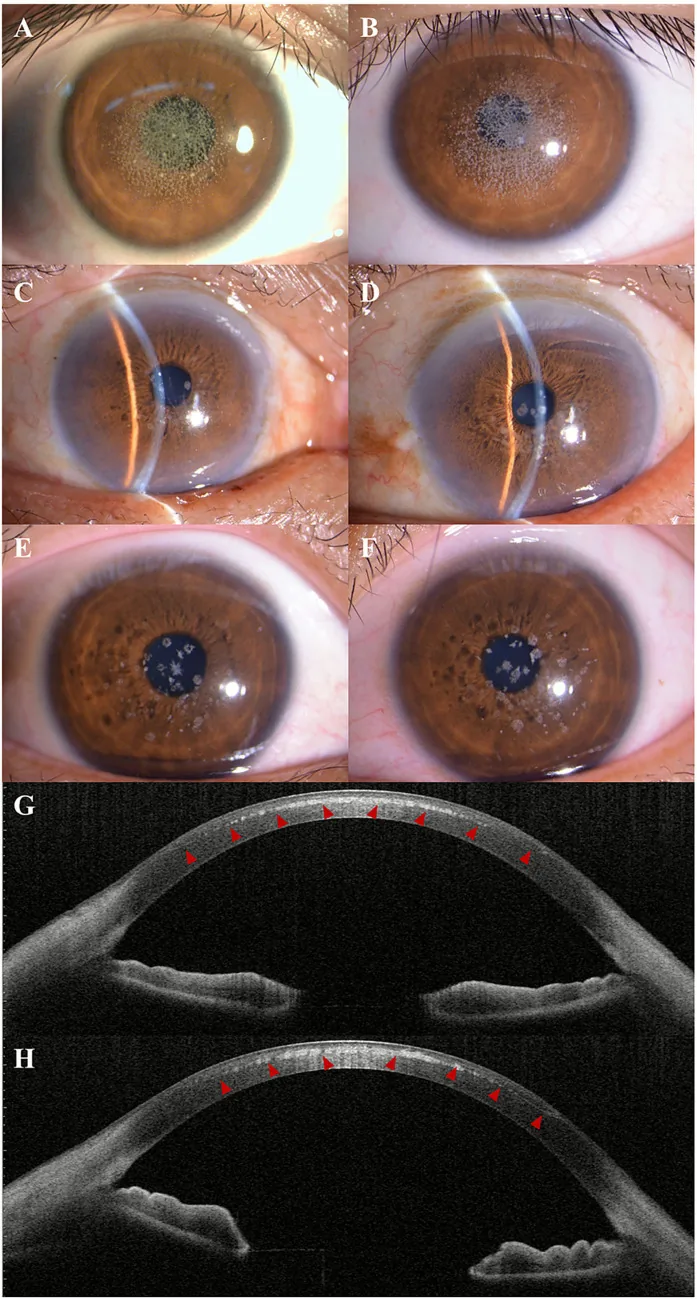
Kuang L, et al. Case Report: Post-LASIK exacerbation of granular corneal dystrophy type 2: a familial case with TGFBI mutation. Front Med (Lausanne). 2025. Figure 1. PMCID: PMC12722859. License: CC BY.
Sur la photographie à la lampe à fente, des opacités granulaires blanc-grisâtre sont dispersées et regroupées du centre à la région paracentrale de la cornée. L’AS-OCT montre des dépôts hyperréflectifs dans le stroma cornéen antérieur, illustrant les signes cliniques de la dystrophie cornéenne granulaire.
Asymptomatique à léger : Chez les hétérozygotes, aux stades précoces à intermédiaires, il n’y a pas de baisse de vision subjective, et il n’est pas rare que la maladie soit découverte fortuitement lors d’un examen de routine. La plupart des patients ne se plaignent de baisse de vision qu’entre 40 et 50 ans.
Éblouissement et photophobie : Lorsque les opacités atteignent la zone pupillaire, les patients se plaignent d’éblouissement diurne et d’une diminution de la sensibilité au contraste.
Érosions épithéliales récurrentes : Les dépôts endommagent la membrane de Bowman et la membrane basale épithéliale, provoquant des douleurs oculaires aiguës, un larmoiement et une rougeur pendant le sommeil ou au réveil.
Baisse de l’acuité visuelle : Lorsque les zones transparentes entre les dépôts deviennent opaques, la vision diminue progressivement3).
Diminution de la sensibilité au contraste : La sensibilité au contraste diminue souvent avant la baisse de l’acuité visuelle (test de Landolt).
Tendance à la cécité diurne : En raison de l’effet de la lumière diffusée en environnement lumineux, les patients se plaignent d’éblouissement à l’extérieur ou en conduisant.
Difficulté de correction par lunettes ou lentilles : La diffusion par les dépôts ne peut être corrigée par des moyens réfractifs.
Chez les homozygotes, une baisse significative de l’acuité visuelle apparaît dès l’enfance (4-7 ans) et un traitement est nécessaire vers l’âge de 10 ans.
Signes cliniques (observés par le médecin lors de l’examen)
Opacités granuleuses : petites opacités granuleuses blanches à blanc-grisâtre, bien délimitées, dispersées dans la partie centrale de la cornée. Elles sont décrites comme des miettes de pain ou des flocons de neige.
Profondeur : sous-épithéliale et dans le stroma cornéen superficiel. N’atteint pas le limbe.
Substance déposée : hyaline uniquement. Se colore en rouge avec la coloration au trichrome de Masson. Ne contient pas d’amyloïde.
Progression : le nombre de granules augmente avec l’âge et leurs limites deviennent floues.
GCD2 (R124H)
Opacités granuleuses : opacités plus grandes que dans GCD1, blanches à blanc-grisâtre, bien délimitées. Les phénotypes sont variés : en forme de dragée, linéaires, étoilées, en massue, etc.
Type mixte : des opacités linéaires fines en forme de réseau, comme dans la dystrophie cornéenne grillagée, peuvent également être observées.
Substance déposée : à la fois hyaline et amyloïde. Positive à la coloration au trichrome de Masson et au rouge Congo, et apparaît en vert-jaune en microscopie à polarisation.
Progression : après 25-30 ans, des opacités blanches denses en forme de bâtonnets ou d’étoiles s’ajoutent dans le stroma moyen. Des dépôts diffus en nappe dans les couches superficielles deviennent plus importants, ce qui constitue une bonne indication pour la PTK3).
Dans les deux types, les opacités sont situées dans la partie centrale de la cornée et n’atteignent pas la région limbique. Habituellement bilatérales avec peu de différence entre les deux yeux.
Les mutations homozygotes présentent un phénotype nettement différent.
Homozygote GCD1 : opacités réticulaires blanches, presque sans espace, à la même profondeur sous l’épithélium cornéen et dans le stroma superficiel. À un stade avancé, l’iris et la chambre antérieure deviennent invisibles.
Homozygote GCD2 : opacités blanches circulaires denses couvrant toute la cornée sauf la périphérie extrême. Si sévère que la blancheur est visible à l’œil nu, seule la transparence limbique est préservée 3). Les cas homozygotes sont une dystrophie cornéenne réfractaire récidivant dans les 1 à 2 ans après PTK ou greffe de cornée.
QQuelle est la différence d'évolution entre homozygotes et hétérozygotes ?
A
Les homozygotes développent la maladie dans l’enfance (4-7 ans) avec une progression rapide. Des opacités blanches couvrent toute la cornée sans espace, nécessitant une PTK ou une greffe de cornée vers l’âge de 10 ans. Même après chirurgie, la récidive survient en 1 à 2 ans, avec une évolution réfractaire. Les hétérozygotes progressent lentement et conservent généralement une bonne vision jusqu’à 40-50 ans.
La GCD est causée par des mutations ponctuelles du gène TGFBI (chromosome 5q31). Le gène TGFBI code pour la protéine de matrice extracellulaire TGFBIp (kératoépithéline). La TGFBIp mutée a une sensibilité réduite à la dégradation protéolytique et s’accumule sous forme de dépôts insolubles anormaux dans le stroma cornéen1,5,7).
Le groupe des dystrophies cornéennes liées à TGFBI comprend 2,4) :
Dystrophie cornéenne granulaire de type 1 (R555W)
Dystrophie cornéenne granulaire de type 2 (R124H, anciennement Avellino)
Antécédents familiaux : Transmission autosomique dominante ; 50 % des enfants d’une personne atteinte présentent un risque de développer la maladie.
Homozygotie : Les homozygotes pour la même mutation présentent un phénotype plus sévère.
Chirurgie cornéenne : La GCD2 peut progresser rapidement après un traumatisme cornéen. L’opacité s’aggrave notamment après une chirurgie réfractive au laser1,8,9).
Ethnie : La GCD2 est fréquente en Asie de l’Est (Corée, Japon). Un effet fondateur génétique a été suggéré3).
Facteurs environnementaux non élucidés : L’exposition aux UV ou le diabète n’ont pas d’effet direct établi à ce jour.
La GCD est une maladie autosomique dominante à pénétrance élevée. Lorsqu’un cas index est diagnostiqué, 50 % des apparentés au premier degré (parents, frères et sœurs, enfants) peuvent être porteurs de la même mutation. L’identification précoce des porteurs asymptomatiques dans la famille permet d’éviter une future chirurgie réfractive et de planifier un suivi régulier pour surveiller la progression1,5). Une anamnèse familiale minutieuse et, si nécessaire, un test génétique sont fortement recommandés, en particulier chez les jeunes souhaitant une LASIK.
Le diagnostic clinique repose sur l’observation d’opacités granuleuses bien délimitées dans le stroma antérieur à la lampe à fente et sur des antécédents familiaux positifs. Une opacité cornéenne bilatérale (dépôts) sans hyperhémie ni œdème cornéen doit faire suspecter une dystrophie cornéenne3).
Dans le diagnostic différentiel des dystrophies cornéennes, il faut d’abord déterminer si les dépôts sont « bien délimités » ou « diffus »3). Pour les dépôts granuleux bien délimités, on distingue la GCD1 (petits) et la GCD2 (grands) en fonction de leur taille. Dans la GCD2, la sclérose diffuse permet d’observer une opacité diffuse en nappe entre les dépôts granuleux, et cette opacité en nappe est une bonne indication pour la PTK3).
Lampe à fente : Observation directe d’opacités granuleuses blanches bien délimitées. Utilisation de la sclérose diffuse, de la rétro-illumination et de l’éclairage en fente.
Microscopie confocale : présence d’opacités irrégulières, hyperréflectives, en forme de miettes de pain entre l’épithélium et la couche de Bowman.
Microscopie ultrasonique biomicroscopique (UBM) : détecte des granules hyperréflectifs dans le stroma superficiel.
Analyse de la topographie cornéenne : fournit des informations supplémentaires sur la densité de l’opacité.
Test génétique : l’analyse du gène TGFBI est utile pour le diagnostic définitif. Au Japon, il est pris en charge par l’assurance maladie depuis avril 2020 en tant que test génétique pour les dystrophies cornéennes 3).
Photographie du segment antérieur : pour le suivi à long terme, il est important de prendre des photographies de haute qualité du segment antérieur lors de la première visite et à chaque visite de suivi, et de les conserver dans le dossier médical.
GCD1 : dépôts hyalins colorés en rouge par le trichrome de Masson. Ne contient pas d’amyloïde. En microscopie électronique, dépôts en forme de bâtonnets ou de trapèzes.
GCD2 : dépôts à la fois hyalins (positifs au trichrome de Masson) et amyloïdes (positifs au rouge Congo, jaune-vert en lumière polarisée). En microscopie électronique, on observe des dépôts denses aux électrons en forme de bâtonnets et des fibrilles amyloïdes 1,7).
Dystrophie cornéenne grillagée de type 1 (LCD1) : mutation TGFBI R124C. Opacités linéaires et grillagées dues à des dépôts d’amyloïde dans le stroma. Souvent associée à des érosions épithéliales récurrentes3)
Dystrophie cornéenne maculaire (MCD) : mutation du gène CHST6. Hérédité autosomique récessive. Opacification diffuse de toute la cornée
Dystrophie cornéenne de Reis-Bücklers : mutation TGFBI R124L. Opacités en carte géographique de la couche de Bowman
Dystrophie cornéenne tachetée (FCD) : mutation PIP5K3. Petites taches blanches dans tout le stroma, généralement asymptomatiques
QLe test génétique est-il pris en charge par l'assurance maladie ?
A
Depuis avril 2020, le test génétique pour les dystrophies cornéennes est pris en charge par l’assurance maladie. Cependant, seuls les établissements agréés peuvent le réaliser, ce qui limite les centres disponibles. En cas de suspicion clinique ou lors de l’envisagement d’une chirurgie réfractive comme le LASIK, un diagnostic de confirmation par test génétique est recommandé.
Aux stades précoces sans baisse de l’acuité visuelle ni érosions épithéliales récurrentes, aucun traitement n’est nécessaire. Une intervention chirurgicale est envisagée lorsque la baisse de vision atteint la zone pupillaire.
Larmes artificielles : collyre d’hyaluronate de sodium à 0,1 % ou 0,3 %, 4 à 6 fois par jour, pour réduire la sécheresse et l’irritation
Lentilles de contact souples thérapeutiques : en cas d’érosions épithéliales récurrentes, protègent la surface oculaire et favorisent la cicatrisation. Port continu recommandé, avec remplacement régulier
Collyres et pommades antibiotiques : pour prévenir les infections secondaires lors d’érosions épithéliales, collyre de lévofloxacine à 0,5 % 3 à 4 fois par jour, et pommade d’ofloxacine au coucher
Solution saline hypertonique (collyre ou pommade à 5% de chlorure de sodium) : peut être utilisée comme traitement adjuvant pour réduire l’œdème épithélial.
Homozygote GCD2 : récidive environ 18 mois après la première PTK, puis environ 3 mois après la deuxième, troisième, etc.1)
Hétérozygote GCD2 : récidive après PTK relativement lente, en moyenne 38,4 mois1)
Utilisation concomitante de mitomycine C (MMC) : l’utilisation de MMC lors de la PTK n’est pas recommandée. En effet, la MMC induit l’apoptose des kératocytes du stroma cornéen, réduisant les cellules responsables de la résorption et de la dégradation de la TGFBIp, ce qui peut accélérer la récidive1)
Cas d’aggravation après LASIK : la PTK est possible, mais l’efficacité est plus élevée après ablation du volet LASIK1,8)
Soins postopératoires : après PTK, utiliser des collyres antibiotiques (lévofloxacine 0,5 %) et des collyres corticostéroïdes (fluorométholone 0,1 %) 4 fois par jour jusqu’à la guérison épithéliale, puis réduire la dose. La guérison épithéliale prend généralement 3 à 5 jours.
Réalisation de la DALK (kératoplastie lamellaire antérieure profonde)
La DALK est une technique qui préserve l’endothélium. On utilise la technique de la grosse bulle (big bubble) pour disséquer et retirer le stroma jusqu’à la membrane de Descemet, puis on suture le stroma du donneur. Comme il n’y a pas de risque de rejet endothélial, le pronostic à long terme est considéré comme meilleur que celui de la PK3). Selon une étude de Kitazawa et al., l’acuité visuelle à 5 ans après DALK pour les dystrophies cornéennes liées à la TGFBI (y compris les formes granuleuse et réticulée) est généralement bonne, et le taux de survie du greffon est élevé10). Au Japon, cette intervention est réalisable dans le cadre de l’assurance maladie.
Arbre décisionnel pour le choix de la technique chirurgicale
Le choix du traitement chirurgical pour la GCD est examiné dans l’ordre suivant1,6).
Dépôts limités au stroma antérieur (sous-épithélial jusqu’à environ 150 μm) → PTK
Récidive après plusieurs PTK ou opacités profondes (150 à 400 μm) → DALK
Opacités sur toute l’épaisseur, atteinte endothéliale associée, ou cas où la DALK n’est pas possible → PK
Cas réfractaires avec récidives répétées après toute technique (en particulier homozygotes GCD2) → envisager une approche de recherche telle que la thérapie génique
Contre-indications de la chirurgie réfractive au laser
La GCD est une contre-indication pour le LASIK, le LASEK, la PRK et le SMILE. Après l’intervention, l’opacité cornéenne s’aggrave rapidement, entraînant une baisse sévère de l’acuité visuelle1,8,9). Après le LASIK, de nombreux petits dépôts granulaires se forment entre le volet et le lit stromal. L’aggravation est plus sévère et l’acuité visuelle finale est moins bonne après le LASIK qu’après la PRK 1,8). Des rapports de cas en Corée et au Japon décrivent de nombreux patients asymptomatiques avant l’intervention qui ont développé une opacité cornéenne marquée quelques mois à quelques années après le LASIK, nécessitant une PTK ou une greffe de cornée8,9).
QQue se passe-t-il si la GCD est découverte après avoir subi un LASIK ?
A
Dans les cas de GCD survenant après un LASIK, des dépôts granulaires se forment rapidement entre le volet et le lit stromal. Les options de traitement comprennent la PTK après retrait du volet LASIK, la DALK et la PK, à considérer dans cet ordre. Une consultation précoce chez un ophtalmologiste est importante.
Le gène TGFBI code pour la protéine de la matrice extracellulaire TGFBIp (kéatoépithéline, 68 kDa). La TGFBIp est impliquée dans l’adhésion, la migration et la prolifération cellulaires, et est également exprimée dans le stroma cornéen normal 1,5,7). Lorsque le gène TGFBI est muté, la TGFBIp mutée devient moins sensible à la dégradation protéolytique et s’accumule sous forme de dépôts insolubles dans le stroma cornéen5,7).
La GCD1 est causée par la mutation Arg555Trp (R555W). La TGFBIp mutée se dépose sous forme de hyaline dans le stroma cornéen superficiel. L’amyloïde n’est pas impliqué 3).
La GCD2 est presque exclusivement causée par la mutation Arg124His (R124H) 1,5). Dans la GCD2, la hyaline et l’amyloïde se déposent tous deux.
Dysfonctionnement de l’autophagie : Un dysfonctionnement de l’autophagie a été rapporté dans la GCD2, ce qui réduit la dégradation de la TGFBIp et favorise son accumulation 1,5)
Dysfonctionnement mitochondrial : Il a été suggéré que la TGFBIp mutée elle-même affecte les fibroblastes cornéens et pourrait provoquer un dysfonctionnement mitochondrial 1)
Effet des néovascularisations cornéennes : Dans les zones de néovascularisation cornéenne, les dépôts ont tendance à diminuer ou à être résorbés. Cette observation confirme le mécanisme par lequel les dépôts se concentrent dans la partie centrale de la cornée, dépourvue de vascularisation 1).
Après LASIK, la TGFBIp se dépose rapidement entre le flap et le lit stromal. On pense que cela est dû à l’intervention chirurgicale dans la zone centrale de la cornée, qui favorise l’accumulation de TGFBIp mutée 1,8). Le fait que les incisions cornéennes lors de la chirurgie de la cataracte (près du limbe) n’aggravent pas la maladie suggère que la distance par rapport au limbe vascularisé est un facteur pertinent 1). Les observations pathologiques d’Awwad et al. suggèrent que les dépôts formés après LASIK s’accumulent avec la TGFBIp lors de l’activation des kératocytes associée à la réaction de cicatrisation à l’interface flap-lit stromal 8).
Les dépôts de GCD1 sont observés en microscopie optique comme des substances acidophiles homogènes, colorées en rouge par le trichrome de Masson. En microscopie électronique, ils apparaissent comme des structures de haute densité électronique en forme de bâtonnets ou de trapèzes, de 100 à 500 nm de diamètre 6).
Dans la GCD2, en plus des dépôts hyalins, on observe des fibrilles amyloïdes (diamètre 8-10 nm). Les fibrilles amyloïdes se colorent en rouge orangé par le rouge Congo et présentent une biréfringence vert pomme en microscopie à lumière polarisée 6). Cette double caractéristique de coloration est utile pour le diagnostic pathologique définitif de la GCD2.
Selon l’analyse protéomique de Poulsen et al., la TGFBIp mutée R124H dans la cornée des patients atteints de GCD2 est moins sensible au clivage par les enzymes protéolytiques que la TGFBIp normale, et des fragments C-terminaux spécifiques s’accumulent sélectivement 6). Il a été suggéré que cette résistance au clivage pourrait être à la base de la formation de fibres hyalines et amyloïdes.
Il a été rapporté que le chlorure de lithium réduit la production de la protéine TGFBI. La thérapie combinée de mélatonine et de rapamycine pourrait inhiber l’expression de la protéine TGFBI tout en activant l’autophagie pour favoriser la dégradation de la TGFBIp mutée 1,5).
Le silençage de l’expression du TGFBI mutant par de petits ARN interférents (siRNA) ou des ARN en épingle à cheveux courts (shRNA) est étudié en phase préclinique. La technologie d’édition génomique CRISPR/Cas9 est également candidate, mais les effets hors cible non intentionnels sur les allèles normaux ou d’autres gènes constituent un défi 1,5).
Électrolyse cornéenne : une utilisation expérimentale a été rapportée pour les cas de récidive après greffe de cornée. Les résultats à long terme sont inconnus.
Aide au diagnostic par apprentissage automatique : le développement d’un modèle d’IA pour identifier automatiquement la GCD à partir de photographies du segment antérieur a été rapporté.
Thérapie par chaperon : des recherches fondamentales sur des chaperons chimiques (acide 4-phénylbutyrique, etc.) qui aident au repliement correct de la TGFBIp mutante sont en cours3).
Feuille épithéliale cornéenne dérivée de cellules iPS : la transplantation d’une feuille épithéliale cornéenne fabriquée à partir de cellules iPS du patient est étudiée comme option future.
Au Japon, des discussions se poursuivent au sein de la Société japonaise d’ophtalmologie et de la Société japonaise de la cornée pour la création d’un registre national des dystrophies cornéennes liées à TGFBI. Depuis la prise en charge par l’assurance maladie du test génétique en 2020, le nombre de cas diagnostiqués génétiquement a augmenté, et des données de suivi à long terme sur les schémas de mutation et les phénotypes spécifiques aux Japonais sont en cours de collecte3,11). À l’avenir, on espère la mise en place d’un système de soins fondé sur des preuves, allant du dépistage des porteurs à la décision d’indication de la chirurgie réfractive.
Chez les patients atteints de GCD, les conseils suivants sont importants.
Protection contre les UV : porter des lunettes de soleil ou des verres avec protection UV lors des sorties pour réduire l’exposition aux UV de la cornée.
Éviter les traumatismes oculaires : les contusions ou les corps étrangers peuvent provoquer des érosions épithéliales récurrentes. Porter des lunettes de protection lors des sports ou du travail.
Consultations régulières : même les hétérozygotes doivent subir un examen à la lampe à fente et un test de vision une fois par an.
Examen familial : recommander un dépistage familial aux parents au premier degré.
Lentilles de contact : les lentilles souples sont généralement possibles, mais le port doit être court et le remplacement régulier strict.
Éviter la chirurgie réfractive : les chirurgies de type LASIK sont absolument contre-indiquées. Choisir une correction par lunettes ou lentilles de contact.
Chang MS, Jun I, Kim EK. Mini-Review: Clinical Features and Management of Granular Corneal Dystrophy Type 2. Korean journal of ophthalmology : KJO. 2023;37(4):340-347. doi:10.3341/kjo.2023.0032. PMID:37336511; PMCID:PMC10427907.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Lisch W, Weiss JS. Clinical and genetic update of corneal dystrophies. Experimental eye research. 2019;186:107715. doi:10.1016/j.exer.2019.107715. PMID:31301286.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern®. Ophthalmology. 2024;131(4):P1-P79.
Poulsen ET, Dyrlund TF, Runager K, et al. Proteomics of granular corneal dystrophy type 2 reveals altered TGFBIp proteolytic processing. J Proteome Res. 2014;13(10):4659-4667.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Awwad ST, Di Pascuale MA, Hogan RN, Forstot SL, McCulley JP, Cavanagh HD. Avellino corneal dystrophy worsening after laser in situ keratomileusis: further clinicopathologic observations and proposed pathogenesis. American journal of ophthalmology. 2008;145(4):656-61. doi:10.1016/j.ajo.2007.12.008. PMID:18243154; PMCID:PMC2661203.
Jun RM, Tchah H, Kim TI, et al. Avellino corneal dystrophy after LASIK. Ophthalmology. 2004;111(3):463-468.
Kitazawa K, Kawasaki S, Shinomiya K, et al. Safety of anterior lamellar keratoplasty for recurrent TGFBI corneal dystrophy. Br J Ophthalmol. 2016;100(4):448-454.