เป็นโรคกระจกตา เสื่อมที่ถ่ายทอดทางพันธุกรรมแบบ autosomal dominant เกิดจากการกลายพันธุ์ของยีน TGFBI ทำให้เกิดการสะสมของไฮยาลินและอะไมลอยด์ในชั้นสโตรมาของกระจกตา
แบ่งเป็นชนิดที่ 1 (การกลายพันธุ์ R555W, Groenouw type 1) และชนิดที่ 2 (การกลายพันธุ์ R124H, เดิมเรียกว่า Avellino corneal dystrophy)
ในญี่ปุ่น ชนิดที่ 2 พบได้บ่อยกว่ามาก ความชุกในเกาหลีประมาณ 11.5 คนต่อประชากร 10,000 คน1)
จากการศึกษาการวินิจฉัยทางพันธุกรรมของมหาวิทยาลัยยามากูจิ โรคกระจกตา เสื่อมหลัก 4 ชนิด (granular type I/II, lattice type I/IIIA, gelatinous drop-like, macular) คิดเป็นประมาณ 96% ของทั้งหมด - ผู้ป่วย homozygous จะมีอาการตั้งแต่วัยเด็ก (อายุ 4-7 ปี) และรุนแรง ส่วนผู้ป่วย heterozygous จะดำเนินโรคอย่างช้าๆ
การรักษาทางเลือกแรกคือ PTK (phototherapeutic keratectomy) หากมีความขุ่นลึกให้เลือก DALK
การผ่าตัดแก้ไขสายตาด้วยเลเซอร์ เช่น LASIK , PRK, LASEK, SMILE เป็นข้อห้าม โรคกระจกตา เสื่อมชนิด Granular (granular corneal dystrophy: GCD ) เป็นโรคกระจกตา ทางพันธุกรรมที่มีลักษณะการสะสมของสารคล้ายเม็ดในชั้นสโตรมาของกระจกตา ตามการจำแนกโรคกระจกตา เสื่อมระหว่างประเทศครั้งที่ 2 (IC3D 2015) จัดอยู่ในกลุ่ม epithelial-stromal TGFBI-related dystrophy2)
สาเหตุเกิดจากการกลายพันธุ์แบบจุดของยีน TGFBI (โครโมโซม 5q31) ซึ่งถ่ายทอดทางพันธุกรรมแบบ autosomal dominant ความแตกต่างของการกลายพันธุ์ทำให้แบ่งเป็น 2 ชนิดดังนี้
การจำแนก การกลายพันธุ์หลัก ชื่ออื่น/ชื่อเดิม สารที่สะสมหลัก GCD 1Arg555Trp (R555W) Granular แบบคลาสสิก, Groenouw type 1 ไฮยาลินเท่านั้น GCD 2Arg124His (R124H) Avellino corneal dystrophy ไฮยาลิน + อะไมลอยด์
GCD 2 ถูกแยกเป็นชนิดย่อยจาก GCD 1 ในปี 1988 และถูกเรียกว่า Avellino corneal dystrophy เนื่องจากครอบครัวแรกที่พบมีต้นกำเนิดจากแคว้นอาเวลลิโนในอิตาลี 1) ต่อมาในปี 1997 ยีน TGFBI ที่เป็นสาเหตุถูกระบุ และพบการกลายพันธุ์ R124H 3) ตั้งแต่การจำแนก IC3D ฉบับที่ 2 (ปี 2015) เป็นต้นมา โรคนี้ได้รับการตั้งชื่ออย่างเป็นทางการว่า granular corneal dystrophy type 2 (GCD 2) และชื่อ “Avellino” ถูกระบุเป็นชื่อทางประวัติศาสตร์ควบคู่กัน 2)
ในระดับสากล กระจกตา เสื่อมชนิดเม็ดเล็ก (granular corneal dystrophy) จัดอยู่ในกลุ่ม epithelial-stromal dystrophies กลุ่มนี้ประกอบด้วยโรคที่เกี่ยวข้องกับ TGFBI 6 โรค ได้แก่ GCD 1, GCD 2, lattice dystrophy (LCD1, LCD3A), Reis-Bücklers และ Thiel-Behnke ซึ่งทั้งหมดเกิดจากการกลายพันธุ์แบบจุดที่แตกต่างกันบนยีน TGFBI บนโครโมโซม 5q31 2,4) ในจักษุวิทยาคลินิกของญี่ปุ่น มีธรรมเนียมเรียกโรคทั้ง 6 นี้รวมกันว่า “TGFBI-associated corneal dystrophies”
Granular corneal dystrophy ถูกบันทึกครั้งแรกโดย Groenouw ในปี 1890 และในขณะนั้นเรียกง่ายๆ ว่า “Groenouw type 1” ในปี 1938 มีการแยกความแตกต่างจาก lattice dystrophy อย่างชัดเจน และเป็นเวลานานที่โรคนี้ถูกถือเป็นโรคเดียวคือ “granular corneal dystrophy” ในปี 1988 มีรายงานรูปแบบโรคที่มีลักษณะทั้งแบบเม็ดเล็กและแบบตาข่ายในครอบครัวจากแคว้นอาเวลลิโน ประเทศอิตาลี ซึ่งต่อมาถูกแยกเป็น GCD 2 (ชนิด Avellino) 1,2) การปรับปรุงการจำแนก IC3D ในปี 2015 ได้สร้างระบบการจำแนกปัจจุบัน และการตั้งชื่อตามจีโนไทป์กลายเป็นมาตรฐานสากล 2)
รูปแบบการถ่ายทอดทางพันธุกรรม : ถ่ายทอดแบบออโตโซมเด่น มีการแทรกซึมสูงชนิดที่ 1 : พบบ่อยในยุโรปและอเมริกา พบน้อยในญี่ปุ่นชนิดที่ 2 : พบมากที่สุดในเอเชียตะวันออก เช่น ญี่ปุ่นและเกาหลี ความชุกในเกาหลีประมาณ 11.5 คนต่อประชากร 10,000 คน 1) สัดส่วนใน TGFBI-associated corneal dystrophies : GCD 2 คิดเป็น 72-91% ในเกาหลีและญี่ปุ่น, 36% ในสหรัฐอเมริกา, และ 3% ในโปแลนด์ 1) ข้อมูลการวินิจฉัยทางพันธุกรรมในญี่ปุ่น : ที่มหาวิทยาลัยยามากูจิ ในช่วง 21 ปีระหว่างปี 2000-2021 มีผู้ป่วยโรคกระจกตา เสื่อม 234 รายที่ได้รับการวินิจฉัยทางพันธุกรรม โดยโรคกระจกตา เสื่อมหลักสี่ชนิด (ชนิดเม็ดละเอียดชนิด I และ II, ชนิดร่างแหชนิด I และ IIIA, ชนิดคล้ายหยดวุ้น, และชนิดเป็นหย่อม) คิดเป็นประมาณ 96% ของทั้งหมด - ลักษณะเฉพาะในเอเชียตะวันออก : โรคกระจกตาเสื่อมชนิดเม็ด ละเอียดในเอเชียตะวันออกส่วนใหญ่เป็นชนิดที่ 2 (R124H) อย่างท่วมท้น - อายุที่เริ่มมีอาการ : ใน heterozygous GCD 2 จะพบความขุ่นเล็กน้อยที่สังเกตได้เฉพาะด้วยกล้อง slit lamp ตั้งแต่วัยเรียน แต่ไม่มีอาการทางตา การมองเห็น ลดลงโดยมีอาการมักปรากฏในช่วงอายุ 40-50 ปีความแตกต่างทางเพศ : เป็นโรคถ่ายทอดทางพันธุกรรมแบบ autosomal dominant จึงไม่มีความแตกต่างทางเพศ
Q
“เม็ดละเอียด” หมายถึงลักษณะใด?
A
หมายถึงภาวะที่มีก้อนเล็กๆ สีขาวถึงขาวเทา ขอบเขตชัดเจน (ตะกอนแบบเม็ดละเอียด) จำนวนมากในชั้นสโตรมาส่วนตื้นบริเวณกลางกระจกตา เมื่อสังเกตด้วยกล้อง slit microscope โดยตรง จะมีลักษณะคล้ายเศษขนมปัง เกล็ดหิมะ หรือเม็ดน้ำตาลกรวด ตะกอนเหล่านี้เกิดจากโปรตีน TGFBI ที่กลายพันธุ์
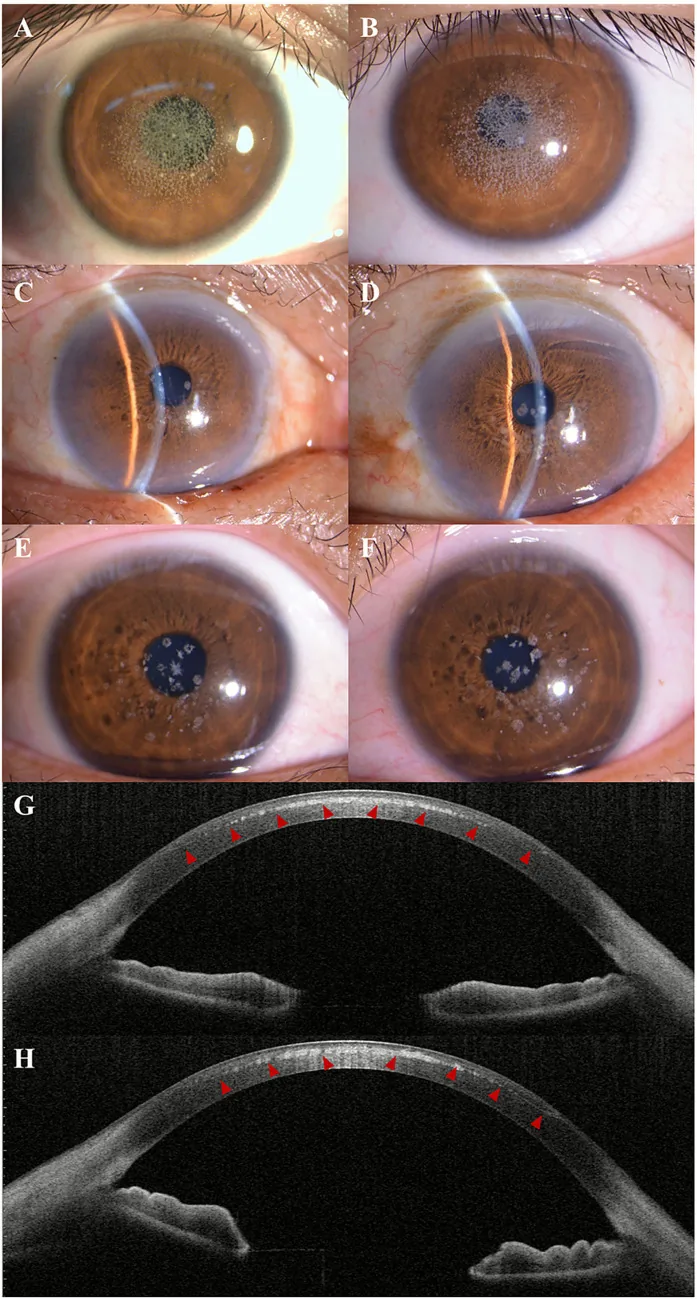
Kuang L, et al. Case Report: Post-LAS
IK exacerbation of granular corneal dystrophy type 2: a familial case with TGFBI mutation. Front Med (Lausanne). 2025. Figure 1. PM
CI D: PMC12722859. License: CC BY.
ภาพถ่าย slit lamp แสดงความขุ่นแบบเม็ดละเอียดสีขาวเทากระจายและรวมกลุ่มกันบริเวณกลางถึงกึ่งกลาง
กระจกตา AS-OCT แสดงตะกอนสะท้อนแสงสูงในชั้นสโตรมาส่วนหน้าของ
กระจกตา ซึ่งเป็นอาการแสดงทางคลินิกของ
โรคกระจกตาเสื่อมชนิดเม็ด ละเอียด
ไม่มีอาการถึงเล็กน้อย : ใน heterozygous ผู้ป่วยระยะแรกถึงระยะกลางมักไม่รู้สึกว่าการมองเห็น ลดลง และมักพบโดยบังเอิญจากการตรวจสุขภาพ หลายรายเริ่มมีอาการมองเห็น ลดลงเมื่ออายุ 40-50 ปีอาการแสบตาและกลัวแสง : เมื่อความขุ่นลามถึงบริเวณรูม่านตา ผู้ป่วยจะรู้สึกแสบตาในเวลากลางวันและการมองเห็น ความแตกต่างลดลงการลอกของเยื่อบุกระจกตา ซ้ำ : ตะกอนทำลายชั้น Bowman และเยื่อฐานเยื่อบุผิว ทำให้เกิดอาการปวดตา อย่างรุนแรง น้ำตาไหล และตาแดง ขณะนอนหลับหรือตื่นนอนการมองเห็น ลดลงการมองเห็น จะลดลงแบบก้าวหน้า3) การมองเห็น ความแตกต่างลดลงการมองเห็น ความแตกต่างมักลดลงก่อนการมองเห็น (การทดสอบด้วยวงแหวน Landolt)แนวโน้มตาบอดกลางวัน : เนื่องจากได้รับผลกระทบจากแสงกระเจิงในที่สว่าง ผู้ป่วยจะรู้สึกแสบตาเมื่ออยู่กลางแจ้งหรือขณะขับรถแก้ไขได้ยากด้วยแว่นตาหรือคอนแทคเลนส์ : การกระเจิงของตะกอนไม่สามารถแก้ไขได้ด้วยการหักเหของแสง
ในโฮโมไซกัส จะมีสายตาลดลงอย่างเห็นได้ชัดตั้งแต่วัยเด็ก (อายุ 4-7 ปี) และจำเป็นต้องได้รับการรักษาเมื่ออายุประมาณ 10 ปี
GCD1 (R555W)
ความขุ่นแบบเม็ด : พบจุดขุ่นสีขาวถึงเทาขนาดค่อนข้างเล็ก ขอบเขตชัดเจน กระจายอยู่บริเวณกลางกระจกตา ลักษณะคล้ายเศษขนมปังหรือเกล็ดหิมะ
ความลึก : อยู่ใต้เยื่อบุกระจกตา และชั้นสโตรมาส่วนตื้น ไม่ลามถึงลิมบัส
สารที่สะสม : มีเฉพาะไฮยาลิน (สารคล้ายแก้ว) เท่านั้น ย้อมสีแดงด้วย Masson’s trichrome ไม่มีอะไมลอยด์
การดำเนินโรค : จำนวนเม็ดขุ่นเพิ่มขึ้นตามอายุ และขอบเขตเริ่มไม่ชัดเจน
GCD2 (R124H)
ความขุ่นแบบเม็ด : เริ่มต้นด้วยจุดขุ่นสีขาวถึงเทา ขอบเขตชัดเจน มีขนาดใหญ่กว่า GCD 1 ลักษณะหลากหลาย เช่น คล้ายลูกอมเม็ดเล็ก เส้นตรง ดาว หรือกระบอง
ชนิดผสม : อาจพบเส้นขุ่นบางๆ คล้ายตาข่าย ซึ่งพบใน lattice corneal dystrophy ร่วมด้วย
สารที่สะสม : ทั้งไฮยาลินและอะไมลอยด์ ย้อมติดสีแดงด้วย Masson’s trichrome และติดสีแดงคองโก ให้สีเขียวอมเหลืองภายใต้กล้องจุลทรรศน์โพลาไรซ์
การดำเนินโรค : เมื่ออายุมากกว่า 25-30 ปี จะมีจุดขุ่นหนาแน่นรูปกระบองหรือดาวเพิ่มขึ้นในชั้นสโตรมาส่วนกลาง ชั้นตื้นจะมีตะกอนแบบกระจายมากขึ้น ซึ่งเป็นข้อบ่งชี้ที่ดีสำหรับ PTK 3)
ในทั้งสองชนิด ความขุ่นจะอยู่บริเวณกลางกระจกตา ไม่ลามถึงขอบลิมบัส โดยปกติเป็นทั้งสองตาและความแตกต่างระหว่างตาซ้ายขวาน้อย
ในผู้ที่มีการกลายพันธุ์แบบโฮโมไซกัส ฟีโนไทป์จะแตกต่างอย่างมาก
GCD 1 โฮโมไซกัสกระจกตา จนถึงชั้นสโตรมาส่วนตื้นในระดับความลึกเดียวกัน โดยแทบไม่มีช่องว่าง เมื่อดำเนินไป ม่านตา และช่องหน้าม่านตา จะไม่สามารถสังเกตเห็นได้GCD 2 โฮโมไซกัสกระจกตา ทั้งหมดยกเว้นบริเวณขอบตาส่วนปลายสุด โดยไม่มีช่องว่าง รุนแรงจนมองเห็นความขาวได้ด้วยตาเปล่า โดยคงความใสเฉพาะบริเวณลิมบัส 3) ผู้ป่วยโฮโมไซกัสเป็นโรคกระจกตา เสื่อมที่ดื้อต่อการรักษา กลับเป็นซ้ำภายในระยะเวลาสั้นๆ 1-2 ปีแม้หลังการทำ PTK หรือการปลูกถ่ายกระจกตา
Q
การดำเนินโรคในโฮโมไซกัสและเฮเทอโรไซกัสแตกต่างกันอย่างไร?
A
โฮโมไซกัสเริ่มมีอาการในวัยเด็ก (4-7 ปี) และดำเนินไปอย่างรวดเร็ว มีความขุ่นสีขาวเต็มพื้นผิวกระจกตา โดยไม่มีช่องว่าง จำเป็นต้องทำ PTK หรือปลูกถ่ายกระจกตา ราวอายุ 10 ปี หลังการผ่าตัดก็กลับเป็นซ้ำภายใน 1-2 ปี ซึ่งเป็นโรคที่ดื้อต่อการรักษา ส่วนเฮเทอโรไซกัสดำเนินไปอย่างช้าๆ โดยทั่วไปสามารถรักษาการมองเห็น ที่ดีได้จนถึงอายุ 40-50 ปี
GCD เกิดจากการกลายพันธุ์แบบจุดในยีน TGFBI (โครโมโซม 5q31) ยีน TGFBI เข้ารหัสโปรตีน TGFBIp (เคอราโตเอพิเทลิน) ซึ่งเป็นโปรตีนเมทริกซ์นอกเซลล์ TGFBIp ที่กลายพันธุ์มีความไวต่อการสลายโปรตีนลดลง จึงสะสมเป็นตะกอนที่ละลายไม่ได้ผิดปกติในชั้นสโตรมาของกระจกตา 1,5,7)
กลุ่มโรคกระจกตา เสื่อมที่เกี่ยวข้องกับ TGFBI ประกอบด้วย2,4) :
Granular corneal dystrophy type 1 (R555W)
Granular corneal dystrophy type 2 (R124H, เดิมชื่อ Avellino)
Lattice corneal dystrophy type 1 (R124C)
Lattice corneal dystrophy type 3A (R501T หรือ L527R)
Reis-Bücklers corneal dystrophy (R124L)
Thiel-Behnke corneal dystrophy (R555Q)
ประวัติครอบครัว : เป็นการถ่ายทอดทางพันธุกรรมแบบออโตโซมเด่น บุตรของผู้ป่วยมีโอกาสเสี่ยงที่จะเกิดโรค 50%โฮโมไซกัส : ผู้ที่เป็นโฮโมไซกัสสำหรับการกลายพันธุ์เดียวกันจะมีฟีโนไทป์ที่รุนแรงกว่าการผ่าตัดตา : GCD 2 สามารถดำเนินไปอย่างรวดเร็วหลังการบาดเจ็บที่กระจกตา โดยเฉพาะหลังการผ่าตัดแก้ไขสายตาด้วยเลเซอร์ ความขุ่นจะเพิ่มขึ้นอย่างชัดเจน1,8,9) เชื้อชาติ : GCD 2 พบได้บ่อยในเอเชียตะวันออก (เกาหลี ญี่ปุ่น) มีการชี้ให้เห็นถึงการมีส่วนร่วมของผลกระทบจากผู้ก่อตั้งทางพันธุกรรม (founder effect)3) ปัจจัยแวดล้อมยังไม่ทราบแน่ชัด : ผลกระทบโดยตรงจากการสัมผัสรังสีอัลตราไวโอเลตหรือโรคเบาหวานยังไม่ได้รับการยืนยันในปัจจุบัน
GCD เป็นโรคที่ถ่ายทอดทางพันธุกรรมแบบออโตโซมเด่นที่มีการแทรกซึมสูง หากผู้ป่วยรายแรกได้รับการวินิจฉัยที่แน่ชัด ญาติสายตรงระดับแรก (พ่อแม่ พี่น้อง บุตร) มีโอกาส 50% ที่จะมีการกลายพันธุ์เดียวกัน การระบุพาหะที่ไม่มีอาการในครอบครัวตั้งแต่เนิ่นๆ จะช่วยให้สามารถวางแผนหลีกเลี่ยงการผ่าตัดแก้ไขสายตาในอนาคต และวางแผนการตรวจติดตามความก้าวหน้าของโรคได้1,5) โดยเฉพาะในวัยรุ่นที่ต้องการทำ LASIK ควรซักประวัติครอบครัวอย่างละเอียดและพิจารณาการตรวจทางพันธุกรรมเมื่อจำเป็น
IK และ PRK
โรคกระจกตาเสื่อมชนิดเม็ด เล็ก (Granular corneal dystrophy) เป็นข้อห้ามในการผ่าตัดแก้ไขสายตาด้วยเลเซอร์ เช่น LASIK , PRK, LASEK, SMILE หลังการผ่าตัด ความขุ่นของกระจกตา อาจแย่ลงอย่างรวดเร็วและทำให้การมองเห็น ลดลงอย่างรุนแรง เมื่อพิจารณาการผ่าตัดแก้ไขสายตาสั้น หรือสายตาเอียง ควรตรวจสอบประวัติครอบครัวและแนะนำให้ตรวจทางพันธุกรรมก่อนการผ่าตัดหากจำเป็น
การวินิจฉัยทางคลินิกขึ้นอยู่กับการสังเกตความขุ่นแบบเม็ดเล็กที่มีขอบเขตชัดเจนในชั้นสโตรมาส่วนหน้าด้วยกล้องจุลทรรศน์ชนิดกรีด (slit lamp) และประวัติครอบครัวที่เป็นบวก หากพบความขุ่นของกระจกตา (การสะสม) ที่เป็นสองตาโดยไม่มีอาการตาแดง หรือกระจกตา บวม ให้สงสัยโรคกระจกตา เสื่อม3)
ในการแยกโรคกระจกตา เสื่อมชนิดต่างๆ ขั้นแรกให้พิจารณาว่าการสะสมนั้นมี “ขอบเขตชัดเจน” หรือ “กระจาย”3) หากเป็นการสะสมแบบเม็ดเล็กที่มีขอบเขตชัดเจน ให้แยก GCD 1 (ขนาดเล็ก) และ GCD 2 (ขนาดใหญ่) ตามขนาดของการสะสม ใน GCD 2 การตรวจด้วยวิธี scleral scatter จะพบความขุ่นแบบแผ่นกระจายระหว่างเม็ดเล็ก ซึ่งความขุ่นแบบแผ่นนี้เป็นข้อบ่งชี้ที่ดีสำหรับการรักษาด้วย PTK 3)
กล้องจุลทรรศน์ชนิดกรีด (slit lamp) : สังเกตความขุ่นแบบเม็ดเล็กสีขาวที่มีขอบเขตชัดเจนโดยตรง ใช้เทคนิค scleral scatter, retroillumination และ transillumination ร่วมด้วยเครื่องตรวจวัดชั้นหน้าด้วยแสง (AS-OCT ) : แสดงความขุ่นสะท้อนแสงสูงในเนื้อกระจกตา ชั้นหน้า มีประโยชน์ในการวางแผนความลึกของการตัดด้วย PTK กล้องจุลทรรศน์คอนโฟคอล ชั้นโบว์แมน กล้องจุลทรรศน์อัลตราซาวนด์ชีวภาพ (UBM ) : ตรวจพบเม็ดสะท้อนแสงสูงในเนื้อกระจกตา ชั้นตื้นการวิเคราะห์รูปทรงกระจกตา การตรวจทางพันธุกรรม : การวิเคราะห์ยีน TGFBI มีประโยชน์ในการวินิจฉัยที่แน่นอน ในญี่ปุ่น ตั้งแต่เดือนเมษายน พ.ศ. 2563 การตรวจยีนจอประสาทตา เสื่อมได้รับการบรรจุในประกันสุขภาพ3) การถ่ายภาพส่วนหน้าของตา : เพื่อการติดตามระยะยาว ควรถ่ายภาพส่วนหน้าคุณภาพสูงทั้งในครั้งแรกและทุกครั้งที่ติดตามผล และบันทึกไว้ในเวชระเบียน
ในการตรวจด้วยกล้องจุลทรรศน์ร่องกราดที่มีความชำนาญ ควรใช้วิธีการตรวจต่อไปนี้ร่วมกันเพื่อเพิ่มความไว3)
การส่องสว่างโดยตรง : ประเมินขนาดและความหนาแน่นของตะกอนเม็ดที่มีขอบเขตชัดเจนการกระเจิงแสงจากตาขาว : เน้นความขุ่นแบบกระจายเป็นแผ่นระหว่างตะกอน มีประโยชน์โดยเฉพาะใน GCD 2การส่องสว่างย้อนกลับ (retro-illumination) : ใช้แสงที่ผ่านบริเวณรูม่านตา เพื่อตรวจหาตะกอนขนาดเล็กการส่องผ่าน (transillumination) : ตรวจพบความขุ่นแบบเม็ดเล็กน้อยใต้เยื่อบุผิวในระยะเริ่มต้น
GCD 1GCD 21,7)
โรคกระจกตา ลายตาข่ายชนิดที่ 1 (LCD1) : การกลายพันธุ์ TGFBI R124C มีการสะสมของอะไมลอยด์ในชั้นสโตรมาทำให้เกิดความขุ่นเป็นเส้นและลายตาข่าย มักเกิดการหลุดลอกของเยื่อบุกระจกตา ซ้ำๆ 3) โรคกระจกตา ขุ่นแบบจุด (MCD ) : การกลายพันธุ์ของยีน CHS T6 ถ่ายทอดทางพันธุกรรมแบบด้อย กระจกตา ทั้งหมดขุ่นแบบกระจายโรคกระจกตา Reis-Bücklers : การกลายพันธุ์ TGFBI R124L ความขุ่นแบบแผนที่ในชั้น Bowmanโรคกระจกตา Thiel-Behnke : การกลายพันธุ์ TGFBI R555Q ความขุ่นแบบรังผึ้งโรคกระจกตา Schnyder : การกลายพันธุ์ UBIAD1 ความขุ่นของกระจกตา จากการสะสมของไขมันโรคกระจกตา ขุ่นแบบจุดเล็ก (FCD) : การกลายพันธุ์ PIP5K3 มีจุดสีขาวเล็กๆ ทั่วสโตรมา มักไม่มีอาการ
Q
การตรวจทางพันธุกรรมสามารถใช้สิทธิประกันสุขภาพได้หรือไม่?
A
ตั้งแต่เดือนเมษายน พ.ศ. 2563 การตรวจทางพันธุกรรมสำหรับโรคกระจกตา เสื่อมได้รับการบรรจุในสิทธิประกันสุขภาพ อย่างไรก็ตาม ต้องมีการรับรองสถานพยาบาล ดังนั้นสถานพยาบาลที่สามารถตรวจได้จึงมีจำกัด หากมีข้อสงสัยจากอาการทางคลินิก หรือกำลังพิจารณาการผ่าตัดแก้ไขสายตา เช่น LASIK ควรยืนยันการวินิจฉัยด้วยการตรวจทางพันธุกรรม
ในระยะแรกที่ยังไม่มีการสูญเสียการมองเห็น หรือการหลุดลอกของเยื่อบุกระจกตา ซ้ำๆ ไม่จำเป็นต้องรักษา เมื่อการมองเห็น ลดลงจนถึงบริเวณรูม่านตา ควรพิจารณาการผ่าตัด
น้ำตาเทียม : ใช้โซเดียมไฮยาลูโรเนต 0.1% หรือ 0.3% หยดตา 4-6 ครั้งต่อวัน เพื่อลดความแห้งและการระคายเคืองคอนแทคเลนส์ชนิดนิ่มเพื่อการรักษา : สำหรับการหลุดลอกของเยื่อบุกระจกตา ซ้ำๆ ช่วยปกป้องผิวตาและส่งเสริมการหาย ควรใส่ตลอดวันและเปลี่ยนเป็นประจำยาหยอดตาหรือยาทาตาต้านเชื้อแบคทีเรีย : เพื่อป้องกันการติดเชื้อซ้ำซ้อนเมื่อมีการหลุดลอกของเยื่อบุกระจกตา ใช้เลโวฟลอกซาซิน 0.5% หยดตา 3-4 ครั้งต่อวัน และใช้ยาทาตาโอฟลอกซาซินก่อนนอนน้ำเกลือความเข้มข้นสูง (ยาหยอดตาหรือยาทาตาโซเดียมคลอไรด์ 5%) : อาจใช้เป็นการรักษาเสริมเพื่อลดอาการบวมของเยื่อบุผิว
เลือกวิธีการรักษาตามความลึกของการสะสม เป้าหมายสูงสุดคือการชะลอการปลูกถ่ายกระจกตา ให้นานที่สุด1,6)
PTK (การตัดเนื้อเยื่อกระจกตาด้วยเลเซอร์เพื่อการรักษา)
ข้อบ่งชี้ : ความขุ่นในชั้นสโตรมาส่วนหน้า แนะนำให้เป็นทางเลือกแรก1)
ข้อดี : สามารถทำซ้ำได้ ไม่มีความเสี่ยงต่อการปฏิเสธ graft ความขุ่นแบบกระจายใต้เยื่อบุผิวเป็นข้อบ่งชี้ที่ดีสำหรับ PTK 3)
เทคนิค : ใช้ excimer laser ตัดสโตรมาประมาณ 50 ไมครอนต่อครั้ง ความลึกของการตัดถูกจำกัดโดยความหนาของกระจกตา 1,8)
ข้อจำกัด : การตัดแต่ละครั้งทำให้เกิดสายตายาว ประมาณ 1.5 D จำนวนครั้งที่ทำได้มีจำกัด
DALK/ALK (การปลูกถ่ายกระจกตาชั้นลึกแบบบางส่วน)
ข้อบ่งชี้ : ความขุ่นในสโตรมาชั้นลึก เมื่อ PTK ไม่สามารถทำได้เนื่องจากข้อจำกัดด้านความหนาของกระจกตา
ข้อดี : DALK เป็นที่นิยมเนื่องจาก endothelial ของกระจกตา ปกติ ไม่มีความเสี่ยงต่อการปฏิเสธแบบ endothelial3)
การกลับเป็นซ้ำ : เกิดซ้ำที่รอยต่อระหว่าง host-graft หรือชั้นสโตรมาส่วนบนของ graft3)
PK (การปลูกถ่ายกระจกตาแบบเต็มชั้น)
ข้อบ่งชี้ : ทางเลือกสุดท้ายเมื่อมีการกลับเป็นซ้ำบ่อยครั้งและมีการสะสมในชั้นสโตรมาส่วนลึก
ค่ามัธยฐานของการกลับเป็นซ้ำ : PK นานที่สุดที่ 13.7 ปี ส่วน PTK , ALK, DALK อยู่ที่ 2.7-3.7 ปี1)
การมองเห็น ที่ดีที่สุดหลังการผ่าตัด1)
GCD 2 โฮโมไซกัสPTK ครั้งแรก และครั้งที่สอง สาม ขึ้นไป กลับมาเป็นซ้ำภายในประมาณ 3 เดือน 1) GCD 2 เฮเทอโรไซกัสPTK ค่อนข้างช้า โดยเฉลี่ย 38.4 เดือน 1) การใช้ไมโตมัยซิน C (MMC) ร่วม : ไม่แนะนำให้ใช้ MMC ระหว่าง PTK เนื่องจาก MMC เหนี่ยวนำให้เกิดอะพอพโทซิส ของเคอราโทไซต์ในสโตรมาของกระจกตา ลดจำนวนเซลล์ที่ทำหน้าที่ดูดซับและย่อยสลาย TGFBIp ซึ่งอาจเร่งการกลับมาเป็นซ้ำ 1) กรณีที่แย่ลงหลัง LASIK : สามารถทำ PTK ได้ แต่ประสิทธิภาพสูงกว่าหลังการนำแผ่นปิด LASIK ออก 1,8) การดูแลหลังผ่าตัด : หลัง PTK ใช้ยาหยอดตาปฏิชีวนะ (levofloxacin 0.5%) และยาหยอดตาสเตียรอยด์ (fluorometholone 0.1%) วันละ 4 ครั้งจนกว่าเยื่อบุผิวจะหายดี จากนั้นลดขนาดลง การหายของเยื่อบุผิวมักใช้เวลา 3-5 วัน
DALK เป็นเทคนิคที่รักษาชั้นเอนโดทีเลียมไว้ โดยใช้เทคนิค big bubble เพื่อลอกและเอาสโตรมาออกจนถึงเหนือเยื่อหุ้มเดสเซเมท แล้วเย็บสโตรมาจากผู้บริจาค เนื่องจากไม่มีความเสี่ยงต่อการปฏิเสธชั้นเอนโดทีเลียม การพยากรณ์โรคระยะยาวจึงดีกว่า PK 3) จากการศึกษาของ Kitazawa และคณะ พบว่าการมองเห็น 5 ปีหลัง DALK สำหรับ TGFBI-associated corneal dystrophy (รวมทั้ง granular และ lattice) โดยทั่วไปดี และอัตราการรอดของ graft ก็สูง 10) ในญี่ปุ่นสามารถทำได้ภายใต้การรักษาพยาบาล
การเลือกการผ่าตัดสำหรับ GCD พิจารณาตามลำดับดังนี้ 1,6)
การสะสมจำกัดอยู่ที่สโตรมาส่วนหน้า (ใต้เยื่อบุผิวกระจกตา ถึงประมาณ 150 ไมครอน) → PTK
กลับมาเป็นซ้ำหลัง PTK หลายครั้ง หรือมีความขุ่นในชั้นลึก (150-400 ไมครอน) → DALK
ความขุ่นทั่วทุกชั้น ร่วมกับความเสียหายของชั้นเอนโดทีเลียม หรือกรณีที่ไม่สามารถทำ DALK ได้ → PK
กรณีดื้อยาที่กลับมาเป็นซ้ำซ้ำแล้วซ้ำเล่าหลังการผ่าตัดทุกเทคนิค (โดยเฉพาะ GCD 2 โฮโมไซกัส) → พิจารณาแนวทางการวิจัย เช่น การบำบัดด้วยยีน
GCD เป็นข้อห้ามสำหรับ LASIK , LASEK, PRK และ SMILE ทุกประเภท หลังผ่าตัด กระจกตา จะขุ่นมัวอย่างรวดเร็วและรุนแรง ทำให้การมองเห็น ลดลงอย่างมาก 1,8,9) หลัง LASIK จะเกิดการสะสมของเม็ดเล็กๆ จำนวนมากระหว่าง flap และ stromal bed การเสื่อมสภาพหลัง LASIK รุนแรงกว่า PRK และการมองเห็น สุดท้ายก็แย่กว่า 1,8) รายงานผู้ป่วยจากเกาหลีและญี่ปุ่นระบุว่าผู้ป่วยที่ไม่มีอาการก่อนผ่าตัด เกิดภาวะกระจกตา ขุ่นมัวอย่างชัดเจนภายในไม่กี่เดือนถึงหลายปีหลัง LASIK และต้องได้รับการรักษาด้วย PTK หรือการปลูกถ่ายกระจกตา 8,9)
ผู้ที่ได้รับการวินิจฉัยว่าเป็น Granular Corneal Dystrophy หรือมีประวัติครอบครัว ควรหลีกเลี่ยงการผ่าตัดแก้ไขสายตาด้วยเลเซอร์ เช่น LASIK , PRK, LASEK, SMILE เนื่องจากมีความเสี่ยงที่กระจกตา จะขุ่นมัวอย่างรวดเร็วหลังผ่าตัด หากต้องการแก้ไขสายตา แนะนำให้ตรวจทางพันธุกรรมก่อนผ่าตัด
Q
จะเกิดอะไรขึ้นหากตรวจพบ GCD หลังจากทำ LASIK แล้ว?
A
ในกรณีที่เกิด GCD หลัง LASIK จะเกิดการสะสมของเม็ดอย่างรวดเร็วระหว่าง flap และ stromal bed ทางเลือกในการรักษา ได้แก่ PTK หลังการนำ LASIK flap ออก, DALK และ PK โดยพิจารณาตามลำดับนี้ การพบจักษุแพทย์โดยเร็วเป็นสิ่งสำคัญ
ยีน TGFBI เข้ารหัสโปรตีน TGFBIp (keratoepithelin, 68 kDa) ซึ่งเป็นโปรตีนในเมทริกซ์นอกเซลล์ TGFBIp เกี่ยวข้องกับการยึดเกาะ การเคลื่อนที่ และการเพิ่มจำนวนของเซลล์ และแสดงออกในเนื้อกระจกตา ปกติ 1,5,7) เมื่อเกิดการกลายพันธุ์ในยีน TGFBI โปรตีน TGFBIp ที่กลายพันธุ์จะมีความไวต่อการย่อยสลายลดลง และสะสมเป็นตะกอนที่ไม่ละลายน้ำในเนื้อกระจกตา 5,7)
GCD 1 เกิดจากการกลายพันธุ์ Arg555Trp (R555W) TGFBIp ที่กลายพันธุ์จะสะสมเป็น hyaline ในชั้นเนื้อกระจกตา ตื้น ไม่มี amyloid 3)
GCD 2 เกือบทั้งหมดเกิดจากการกลายพันธุ์ Arg124His (R124H) 1,5) ใน GCD 2 จะมีการสะสมทั้ง hyaline และ amyloid
ความผิดปกติของ autophagy : ใน GCD 2 มีรายงานความผิดปกติของ autophagy ซึ่งทำให้การย่อยสลาย TGFBIp ลดลง ส่งเสริมการสะสม 1,5) ความผิดปกติของไมโทคอนเดรีย : มีข้อเสนอแนะว่า TGFBIp ที่กลายพันธุ์อาจส่งผลต่อไฟโบรบลาสต์ของกระจกตา ทำให้เกิดความผิดปกติของไมโทคอนเดรีย 1) ผลของเส้นเลือดใหม่ที่กระจกตา : ในบริเวณที่มีเส้นเลือดใหม่ที่กระจกตา มักมีแนวโน้มที่ตะกอนจะลดลงหรือถูกดูดซึมกลับ ซึ่งเป็นข้อค้นพบที่สนับสนุนกลไกที่ตะกอนจะรวมตัวกันที่บริเวณกลางกระจกตา ซึ่งไม่มีหลอดเลือดมาเลี้ยง1)
หลังการทำ LASIK จะมีการสะสมของ TGFBIp อย่างรวดเร็วระหว่าง flap และ stromal bed ซึ่งเชื่อว่าเกิดจากการผ่าตัดบริเวณกลางกระจกตา ที่กระตุ้นให้เกิดการสะสมของ TGFBIp ที่กลายพันธุ์1,8) การผ่าตัดต้อกระจก ซึ่งมีแผลที่กระจกตา บริเวณใกล้ลิมบัส (limbus) ไม่ทำให้อาการแย่ลง จึงสันนิษฐานว่าระยะห่างจากลิมบัส ที่มีหลอดเลือดมีความเกี่ยวข้อง1) การสังเกตทางพยาธิวิทยาของ Awwad และคณะชี้ให้เห็นว่าตะกอนที่เกิดขึ้นหลัง LASIK เกิดจากการสะสมของ TGFBIp ร่วมกับการกระตุ้นของเคอราโทไซต์ (keratocyte) ซึ่งเป็นส่วนหนึ่งของกระบวนการสมานแผลที่รอยต่อระหว่าง flap และ stromal bed8)
ตะกอนใน GCD 1 จะถูกสังเกตเห็นเป็นสาร eosinophilic ที่มีลักษณะเป็นเนื้อเดียวกัน (homogeneous) ภายใต้กล้องจุลทรรศน์แบบใช้แสง และจะย้อมติดสีแดงด้วย Masson’s trichrome stain ในระดับกล้องจุลทรรศน์อิเล็กตรอน จะพบเป็นโครงสร้างที่มีความหนาแน่นอิเล็กตรอนสูงรูปแท่งหรือรูปสี่เหลี่ยมคางหมูขนาดเส้นผ่านศูนย์กลาง 100-500 นาโนเมตร6)
ใน GCD 2 นอกจากตะกอนไฮยาลิน (hyaline) แล้ว ยังพบเส้นใยอะไมลอยด์ (amyloid fibrils) ขนาดเส้นผ่านศูนย์กลาง 8-10 นาโนเมตร เส้นใยอะไมลอยด์จะย้อมติดสีส้มแดงด้วย Congo red และแสดงการหักเหสองแนว (birefringence) สีเขียวแอปเปิล (apple green) ภายใต้กล้องจุลทรรศน์โพลาไรซ์6) ลักษณะการย้อมสองแบบนี้มีประโยชน์ในการวินิจฉัยยืนยันทางพยาธิวิทยาของ GCD 2
จากการวิเคราะห์โปรตีโอมิกส์ของ Poulsen และคณะ พบว่าในกระจกตา ของผู้ป่วย GCD 2 TGFBIp ที่กลายพันธุ์ R124H-TGFBIp จะถูกตัดโดยเอนไซม์โปรตีเอสได้ยากกว่า TGFBIp ปกติ และชิ้นส่วน C-terminal ที่เฉพาะเจาะจงจะสะสมอย่างเลือกสรร6) ความต้านทานต่อการตัดนี้เป็นพื้นฐานที่อาจนำไปสู่การเกิดเส้นใยไฮยาลินและอะไมลอยด์
กำลังมีการพัฒนาวิธีการรักษาใหม่สำหรับ GCD 2 แต่ทั้งหมดยังอยู่ในขั้นตอนการวิจัย1,5)
ลิเธียมคลอไรด์ (Lithium chloride) ได้รับรายงานว่าสามารถลดการผลิตโปรตีน TGFBI การใช้เมลาโทนินร่วมกับราปามัยซิน (rapamycin) อาจยับยั้งการแสดงออกของโปรตีน TGFBI และในขณะเดียวกันก็กระตุ้น autophagy เพื่อส่งเสริมการย่อยสลาย TGFBIp ที่กลายพันธุ์1,5)
การใช้ small interfering RNA (siRNA) หรือ short hairpin siRNA (shRNA) เพื่อ silencing การแสดงออกของ TGFBI ที่กลายพันธุ์กำลังอยู่ในระหว่างการศึกษาก่อนทางคลินิก เทคโนโลยีการตัดต่อยีนด้วย CRISPR/Cas9 ก็เป็นตัวเลือกหนึ่ง แต่ยังมีปัญหาเรื่องผลกระทบที่ไม่พึงประสงค์ต่ออัลลีลปกติหรือยีนอื่น ๆ (off-target effects)1,5)
การสลายกระจกตา ด้วยไฟฟ้า (corneal electrolysis) : มีรายงานการใช้ทดลองในผู้ป่วยที่กลับเป็นซ้ำหลังการปลูกถ่ายกระจกตา ผลระยะยาวยังไม่ทราบแน่ชัดการช่วยวินิจฉัยด้วยการเรียนรู้ของเครื่อง : มีรายงานการพัฒนาแบบจำลอง AI ที่สามารถระบุ GCD โดยอัตโนมัติจากภาพถ่ายส่วนหน้าของตาการบำบัดด้วยชาเปอโรน : การวิจัยพื้นฐานเกี่ยวกับชาเปอโรนเคมี (เช่น 4-phenylbutyric acid) ที่ช่วยให้ TGFBIp ที่กลายพันธุ์พับตัวได้อย่างถูกต้องกำลังดำเนินการอยู่3) แผ่นเยื่อบุผิวกระจกตา ที่ได้จากเซลล์ iPS : การปลูกถ่ายแผ่นเยื่อบุผิวกระจกตา ที่สร้างจากเซลล์ iPS ของผู้ป่วยกำลังถูกวิจัยในฐานะทางเลือกในอนาคต
ในประเทศญี่ปุ่น สมาคมจักษุวิทยาญี่ปุ่นและสมาคมกระจกตา ญี่ปุ่นกำลังหารือเกี่ยวกับการสร้างทะเบียนแห่งชาติสำหรับ TGFBI-associated corneal dystrophy ตั้งแต่ปี 2020 ที่การตรวจทางพันธุกรรมได้รับการบรรจุในประกันสุขภาพ จำนวนผู้ป่วยที่ได้รับการวินิจฉัยทางพันธุกรรมเพิ่มขึ้น และข้อมูลการติดตามระยะยาวเกี่ยวกับรูปแบบการกลายพันธุ์และฟีโนไทป์เฉพาะของคนญี่ปุ่นกำลังสะสม3,11) ในอนาคต คาดว่าจะมีการพัฒนาระบบการดูแลทางการแพทย์ตามหลักฐาน ตั้งแต่การคัดกรองพาหะไปจนถึงการตัดสินใจเกี่ยวกับการผ่าตัดแก้ไขสายตา
สำหรับผู้ป่วย GCD คำแนะนำในการดำเนินชีวิตต่อไปนี้มีความสำคัญ
การป้องกันรังสียูวี : สวมแว่นกันแดดหรือแว่นตาที่มีฟังก์ชันป้องกันรังสียูวีเมื่อออกนอกบ้าน เพื่อลดการสัมผัสรังสียูวีที่กระจกตา หลีกเลี่ยงการบาดเจ็บที่ตา : การกระแทกหรือสิ่งแปลกปลอมอาจทำให้เกิดการลอกของเยื่อบุผิวซ้ำ ควรสวมแว่นตาป้องกันระหว่างเล่นกีฬาหรือทำงานการตรวจติดตามเป็นประจำ : แม้ใน heterozygous ควรได้รับการตรวจด้วย slit lamp และวัดสายตาปีละครั้งการตรวจครอบครัว : แนะนำให้ญาติสายตรงระดับแรกได้รับการคัดกรองภายในครอบครัวคอนแทคเลนส์ : โดยทั่วไปสามารถใช้เลนส์นิ่มได้ แต่ควรลดระยะเวลาการใส่และเปลี่ยนเป็นประจำหลีกเลี่ยงการผ่าตัดแก้ไขสายตา : การผ่าตัดแบบ LASIK เป็นข้อห้ามเด็ดขาด เลือกแก้ไขด้วยแว่นตาหรือคอนแทคเลนส์
การรักษาด้วยยาและการบำบัดด้วยยีน ที่กล่าวถึงในบทความนี้ทั้งหมดอยู่ในขั้นตอนการวิจัย และยังไม่มีการนำมาใช้ทางคลินิกในขณะนี้ โปรดปรึกษาจักษุแพทย์ผู้เชี่ยวชาญเพื่อกำหนดแนวทางการรักษา การตรวจทางพันธุกรรมมีสิทธิ์ได้รับการคุ้มครองจากประกันสุขภาพตั้งแต่ปี 2563 ดังนั้นโปรดปรึกษาแพทย์ผู้ดูแลเพื่อการวินิจฉัยที่แน่ชัดและการตรวจคัดกรองครอบครัว
Chang MS , Jun I, Kim EK. Mini-Review: Clinical Features and Management of Granular Corneal Dystrophy Type 2. Korean journal of ophthalmology : KJO. 2023;37(4):340-347. doi:10.3341/kjo.2023.0032. PMID:37336511; PMCI D:PMC10427907.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Lisch W, Weiss JS. Clinical and genetic update of corneal dystrophies. Experimental eye research. 2019;186:107715. doi:10.1016/j.exer.2019.107715. PMID:31301286.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern®. Ophthalmology. 2024;131(4):P1-P79.
Poulsen ET, Dyrlund TF, Runager K, et al. Proteomics of granular corneal dystrophy type 2 reveals altered TGFBIp proteolytic processing. J Proteome Res. 2014;13(10):4659-4667.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Awwad ST, Di Pascuale MA, Hogan RN, Forstot SL, McCulley JP, Cavanagh HD. Avellino corneal dystrophy worsening after laser in situ keratomileusis: further clinicopathologic observations and proposed pathogenesis. American journal of ophthalmology. 2008;145(4):656-61. doi:10.1016/j.ajo.2007.12.008. PMID:18243154; PMCI D:PMC2661203.
Jun RM, Tchah H, Kim TI, et al. Avellino corneal dystrophy after LASIK . Ophthalmology. 2004;111(3):463-468.
Kitazawa K, Kawasaki S, Shinomiya K, et al. Safety of anterior lamellar keratoplasty for recurrent TGFBI corneal dystrophy. Br J Ophthalmol. 2016;100(4):448-454.
日本眼科学会. 角膜ジストロフィ遺伝子検査の保険収載について. 日本眼科学会雑誌. 2020;124:厚生労働省告示に基づく2020年4月1日収載.