دیستروفی قرنیه دانهای (granular corneal dystrophy: GCD) یک بیماری ارثی قرنیه است که با رسوبات دانهای در استرومای قرنیه مشخص میشود. در طبقهبندی بینالمللی دیستروفیهای قرنیه نسخه دوم (IC3D 2015)، در گروه دیستروفیهای اپیتلیال-استرومایی مرتبط با TGFBI طبقهبندی میشود 2).
علت آن جهش نقطهای در ژن TGFBI (کروموزوم 5q31) است و به صورت اتوزومال غالب به ارث میرسد. بر اساس نوع جهش، به دو نوع زیر تقسیم میشود.
GCD2 در سال 1988 به عنوان زیرگروهی مستقل از GCD1 گزارش شد و از آنجایی که اولین خانواده از منطقه آولینو در ایتالیا بود، دیستروفی قرنیه آولینو نامیده شد 1). سپس در سال 1997 ژن عامل TGFBI شناسایی و جهش R124H تعیین شد 3). پس از ویرایش دوم طبقهبندی IC3D (2015)، رسماً دیستروفی قرنیه دانهای نوع 2 (GCD2) نامگذاری شد و «آولینو» به عنوان نام تاریخی در کنار آن ذکر میشود 2).
دیستروفی قرنیه دانهای در سطح بینالمللی در گروه دیستروفیهای اپیتلیال-استرومال طبقهبندی میشود. این گروه شامل شش بیماری مرتبط با TGFBI است: دانهای (GCD1 و GCD2)، مشبک (LCD1 و LCD3A)، Reis-Bücklers و Thiel-Behnke که همگی ناشی از جهشهای نقطهای مختلف در ژن TGFBI روی کروموزوم 5q31 هستند 2,4). در چشمپزشکی بالینی ژاپن، این شش بیماری معمولاً «دیستروفیهای قرنیه مرتبط با TGFBI» نامیده میشوند.
دیستروفی قرنیه دانهای اولین بار در سال 1890 توسط Groenouw گزارش شد و در آن زمان صرفاً «نوع 1 گرونو» نامیده میشد. در سال 1938 تمایز آن از دیستروفی مشبک مشخص شد و مدتها به عنوان یک بیماری واحد با نام «دیستروفی قرنیه دانهای» در نظر گرفته میشد. در سال 1988، در خانوادهای از منطقه آولینو ایتالیا، نوعی بیماری با ویژگیهای هر دو نوع دانهای و مشبک گزارش شد که بعداً به عنوان GCD2 (نوع آولینو) جدا شد 1,2). با بازبینی طبقهبندی IC3D در سال 2015، سیستم طبقهبندی فعلی تثبیت شد و نامگذاری بر اساس ژنوتیپ به استاندارد بینالمللی تبدیل شد 2).
نوع 2: بسیار شایع در شرق آسیا مانند ژاپن و کره. شیوع در کره حدود 11.5 نفر در 10000 نفر 1)
نسبت در میان دیستروفیهای قرنیه مرتبط با TGFBI: GCD2 در کره و ژاپن 72-91٪، در ایالات متحده 36٪، در لهستان 3٪ 1)
دادههای تشخیص ژنتیکی در ژاپن: در دانشگاه یاماگوچی، طی ۲۱ سال از ۲۰۰۰ تا ۲۰۲۱، ۲۳۴ بیمار مبتلا به دیستروفی قرنیه تحت تشخیص ژنتیکی قرار گرفتند و چهار دیستروفی اصلی قرنیه (نوع I و II گرانولار، نوع I و IIIA لاتیس، کلوئیدی قطرهای، و ماکولار) حدود ۹۶٪ از کل موارد را تشکیل دادند. - ویژگی در شرق آسیا: دیستروفی قرنیه گرانولار در شرق آسیا عمدتاً از نوع ۲ (R124H) است. - سن شروع: در هتروزیگوتهای GCD2، از سن مدرسه کدورتهای میکروسکوپی قابل مشاهده با لامپ اسلیت وجود دارد اما علائم ذهنی ندارند. کاهش بینایی ذهنی معمولاً در دهه ۴۰ تا ۵۰ سالگی ظاهر میشود.
تفاوت جنسیتی: این بیماری اتوزومال غالب است و تفاوت جنسیتی ندارد.
Q«گرانولار» به چه وضعیتی اشاره دارد؟
A
به وضعیتی اطلاق میشود که در لایههای سطحی استرومای مرکزی قرنیه، تودههای کوچک سفید تا سفید-خاکستری با مرز مشخص (رسوبات دانهای) به تعداد زیاد ایجاد میشود. با میکروسکوپ لامپ اسلیت، این رسوبات به صورت خرده نان، دانه برف، یا شکر تگرگی توصیف میشوند. رسوبات ناشی از پروتئین TGFBI جهشیافته هستند.
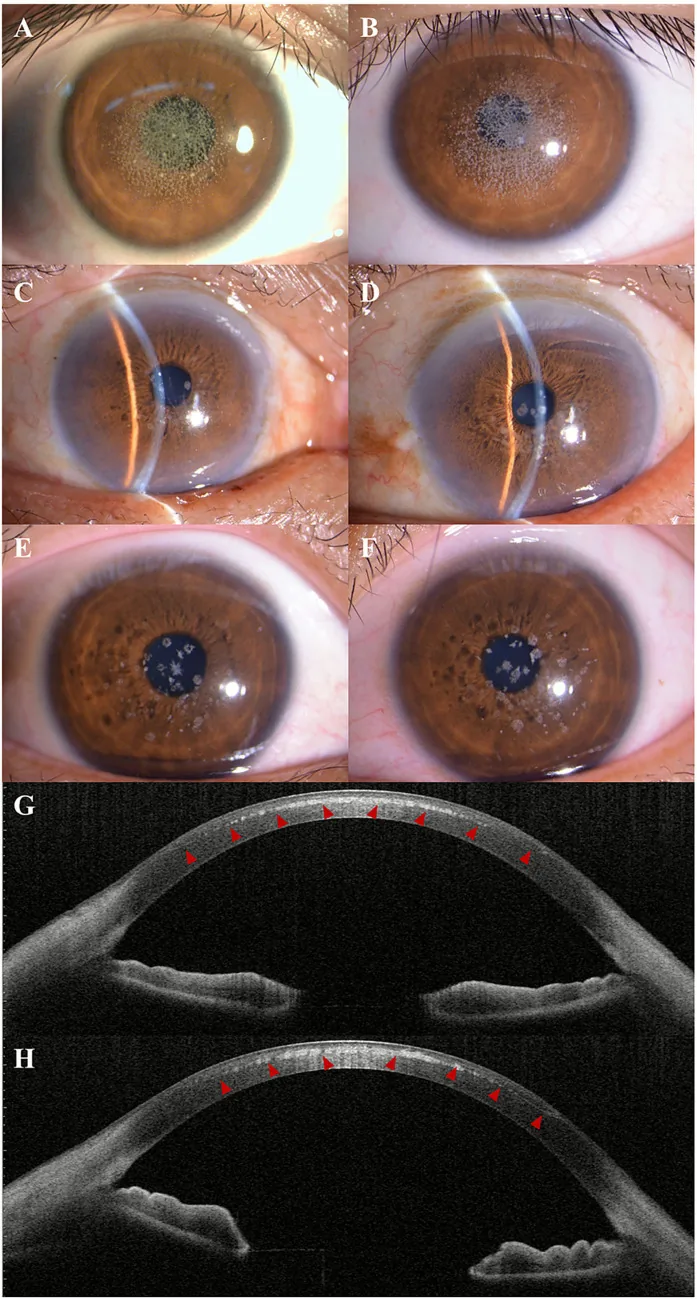
Kuang L, et al. Case Report: Post-LASIK exacerbation of granular corneal dystrophy type 2: a familial case with TGFBI mutation. Front Med (Lausanne). 2025. Figure 1. PMCID: PMC12722859. License: CC BY.
در عکس لامپ اسلیت، کدورتهای دانهای سفید-خاکستری به صورت پراکنده و تجمعیافته در مرکز و اطراف مرکز قرنیه دیده میشود. در AS-OCT، رسوبات با بازتاب بالا در استرومای قدامی قرنیه مشاهده میشود که یافته بالینی دیستروفی قرنیه گرانولار را نشان میدهد.
بدون علامت تا خفیف: در هتروزیگوتها، در مراحل اولیه تا میانی، کاهش بینایی احساس نمیشود و اغلب به طور تصادفی در معاینات کشف میشود. بسیاری از بیماران در دهه ۴۰ تا ۵۰ سالگی از کاهش بینایی شکایت میکنند.
خیرگی و حساسیت به نور: هنگامی که کدورتها به ناحیه مردمک میرسند، بیماران از خیرگی در روز و کاهش حساسیت کنتراست شکایت میکنند.
فرسایش مکرر اپیتلیوم قرنیه: رسوبات به لایه بومن و غشای پایه اپیتلیوم آسیب میرسانند و باعث درد شدید چشم، اشکریزی و قرمزی در هنگام خواب یا بیدار شدن میشوند.
کاهش بینایی: هنگامی که نواحی شفاف بین رسوبات کدر میشوند، بینایی به تدریج کاهش مییابد3).
کاهش حساسیت کنتراست: اغلب قبل از کاهش بینایی (تست حلقه لاندولت)، حساسیت کنتراست کاهش مییابد.
تمایل به شبکوری: به دلیل تأثیر شدید نور پراکنده در محیط روشن، بیماران در فضای باز یا هنگام رانندگی از خیرگی شکایت میکنند.
عدم اصلاح با عینک یا لنز: پراکندگی ناشی از رسوبات با اصلاح انکساری بهبود نمییابد.
در افراد هموزیگوت، کاهش قابل توجه بینایی از دوران کودکی (۴ تا ۷ سالگی) شروع میشود و حدود ۱۰ سالگی نیاز به درمان پیدا میکند.
یافتههای بالینی (یافتههایی که پزشک در معاینه تأیید میکند)
کدورتهای دانهای: کدورتهای دانهای سفید تا خاکستری مایل به سفید، نسبتاً کوچک و با مرز مشخص در مرکز قرنیه پراکنده میشوند. به صورت خرده نان یا دانه برف توصیف میشوند.
عمق: زیر اپیتلیوم قرنیه و لایههای سطحی استرومای قرنیه. به لیمبوس نمیرسد.
ماده رسوبی: فقط هیالین. با رنگآميزی ماسون تریکروم قرمز رنگ میشود. حاوی آمیلوئید نیست.
پیشرفت: با افزایش سن، تعداد دانهها افزایش یافته و مرزها نامشخص میشوند.
GCD2 (R124H)
کدورتهای دانهای: با کدورتهای سفید تا خاکستری مایل به سفید بزرگتر از GCD1 و با مرز مشخص شروع میشود. فنوتیپها متنوع هستند: به شکل آب نبات قند، خطی، ستارهای، چماقی و غیره.
نوع مختلط: گاهی خطوط نازک مشبک مانند دیستروفی قرنیه مشبک نیز دیده میشود.
ماده رسوبی: هم هیالین و هم آمیلوئید. رنگآميزی ماسون تریکروم مثبت و کنگو رد مثبت، در میکروسکوپ پلاریزاسیون رنگ زرد-سبز نشان میدهد.
پیشرفت: در سنین بالای ۲۵ تا ۳۰ سال، کدورتهای سفید ضخیم به شکل چماق و ستاره در لایه میانی استروما اضافه میشود. رسوبات سطحی منتشر و صفحهای افزایش یافته و PTK اندیکاسیون خوبی دارد3).
در هر دو نوع، کدورتها در مرکز قرنیه قرار دارند و به اطراف لیمبوس نمیرسند. معمولاً دوطرفه و با تفاوت کم بین دو چشم هستند.
در جهشهای هموزیگوت، فنوتیپ به طور قابل توجهی متفاوت است.
هموزیگوت GCD1: کدورتهای سفید مشبک تقریباً بدون فاصله در یک عمق، از زیر اپیتلیوم تا لایههای سطحی استروما وجود دارد. با پیشرفت، عنبیه و اتاق قدامی قابل مشاهده نیستند.
هموزیگوت GCD2: کدورتهای سفید دایرهای و متراکم در تمام سطح قرنیه به جز محیطیترین ناحیه ایجاد میشود. آنقدر شدید است که با چشم غیرمسلح نیز سفیدی قابل تشخیص است و تنها شفافیت لیمبوس حفظ میشود3). موارد هموزیگوت یک دیستروفی قرنیه مقاوم به درمان هستند که حتی پس از PTK یا پیوند قرنیه در مدت کوتاه ۱ تا ۲ سال عود میکنند.
Qسیر بیماری در هموزیگوتها و هتروزیگوتها چه تفاوتی دارد؟
A
هموزیگوتها در دوران کودکی (۴ تا ۷ سالگی) شروع میشوند و پیشرفت سریعی دارند. کدورتهای سفید بدون فاصله در تمام سطح قرنیه ایجاد میشود و در حدود ۱۰ سالگی نیاز به PTK یا پیوند قرنیه پیدا میکنند. حتی پس از جراحی نیز در عرض ۱ تا ۲ سال عود میکنند و سیر مقاوم به درمان دارند. هتروزیگوتها به آرامی پیشرفت میکنند و معمولاً تا دهه ۴۰ تا ۵۰ سالگی بینایی خوبی حفظ میشود.
GCD در اثر جهشهای نقطهای در ژن TGFBI (کروموزوم 5q31) ایجاد میشود. ژن TGFBI پروتئین ماتریکس خارج سلولی TGFBIp (کراتواپیسلین) را کد میکند. TGFBIp جهشیافته حساسیت کمتری به تجزیه پروتئینی دارد و به صورت رسوبات غیرقابل حل غیرطبیعی در استرومای قرنیه تجمع مییابد1,5,7).
گروه دیستروفیهای قرنیه مرتبط با TGFBI شامل موارد زیر است2,4):
دیستروفی قرنیه دانهای نوع 1 (R555W)
دیستروفی قرنیه دانهای نوع 2 (R124H، سابقاً Avellino)
GCD یک بیماری اتوزومال غالب با نفوذ بالا است. در صورت تشخیص قطعی در فرد شاخص، ۵۰٪ از بستگان درجه یک (والدین، خواهر و برادر، فرزندان) ممکن است همان جهش را داشته باشند. شناسایی زودهنگام ناقلان بدون علامت در خانواده میتواند به اجتناب از جراحی انکساری در آینده و برنامهریزی ویزیتهای منظم برای پیگیری پیشرفت بیماری کمک کند1,5). به ویژه در جوانانی که متقاضی LASIK هستند، گرفتن شرح حال دقیق خانوادگی و در صورت لزوم انجام آزمایش ژنتیک به شدت توصیه میشود.
تشخیص بالینی بر اساس مشاهده کدورتهای دانهای با مرز مشخص در استرومای قدامی با لامپ شکافی و سابقه خانوادگی مثبت است. در صورت وجود کدورت قرنیه دوطرفه (رسوب) بدون پرخونی و ادم قرنیه، باید به دیستروفی قرنیه مشکوک شد3).
در افتراق دیستروفیهای قرنیه، ابتدا باید مشخص شود که رسوبات «مرز مشخص» دارند یا «منتشر» هستند3). اگر رسوبات دانهای با مرز مشخص باشند، بر اساس اندازه رسوبات، GCD1 (کوچک) از GCD2 (بزرگ) افتراق داده میشود. در GCD2، با روش پراکندگی صلبیه میتوان کدورتهای سطحی منتشر بین رسوبات دانهای را مشاهده کرد که این کدورت سطحی نشانه خوبی برای PTK است3).
آنالیز توپوگرافی قرنیه: اطلاعات اضافی در مورد تراکم کدورت ارائه میدهد.
آزمایش ژنتیکی: آنالیز ژن TGFBI برای تشخیص قطعی مفید است. در ژاپن از آوریل 2020 به عنوان آزمایش ژنتیکی دیستروفی قرنیه تحت پوشش بیمه قرار گرفته است 3).
عکسبرداری از بخش قدامی: برای پیگیری طولانیمدت، گرفتن عکسهای با کیفیت بالا از بخش قدامی در اولین ویزیت و هر ویزیت بعدی و ثبت آن در پرونده پزشکی مهم است.
GCD1: رسوبات هیالین که با رنگآمیز ماسون تریکروم قرمز رنگ میشوند. حاوی آمیلوئید نیست. در میکروسکوپ الکترونی رسوبات میلهای یا ذوزنقهای شکل.
GCD2: هر دو هیالین (مثبت در رنگآمیز ماسون تریکروم) و آمیلوئید (مثبت در رنگآمیز کنگو رد، با میکروسکوپ پلاریزان به رنگ زرد-سبز) رسوب میکنند. در میکروسکوپ الکترونی رسوبات الکتروندنس میلهای و فیبریلهای آمیلوئید مشاهده میشود 1,7).
دیستروفی قرنیهخالدار (FCD): جهش PIP5K3. لکههای سفید کوچک در سراسر استروما، معمولاً بدون علامت
Qآیا آزمایش ژنتیک تحت پوشش بیمه است؟
A
از آوریل 2020، آزمایش ژنتیک دیستروفی قرنیه تحت پوشش بیمه قرار گرفته است. با این حال، به دلیل نیاز به تأیید مرکز، مراکز قادر به انجام آزمایش محدود هستند. در صورت مشکوک بودن یافتههای بالینی یا در نظر گرفتن جراحی انکساری مانند LASIK، تشخیص قطعی با آزمایش ژنتیک توصیه میشود.
در مراحل اولیه که کاهش بینایی یا فرسایش مکرر اپیتلیوم وجود ندارد، نیازی به درمان نیست. زمانی که کاهش بینایی به ناحیه مردمک میرسد، مداخله جراحی در نظر گرفته میشود.
هموزیگوت GCD2: عود حدود 18 ماه پس از اولین PTK رخ میدهد و پس از دومین و سومین بار، حدود 3 ماه بعد عود میکند1)
هتروزیگوت GCD2: عود پس از PTK نسبتاً آهسته است و به طور متوسط 38.4 ماه طول میکشد1)
استفاده همزمان از میتومایسین C (MMC): استفاده از MMC در حین PTK توصیه نمیشود. زیرا MMC باعث آپوپتوز کراتوسیتهای استرومای قرنیه شده و سلولهای مسئول بازجذب و تجزیه TGFBIp را کاهش میدهد که ممکن است عود را تسریع کند1)
موارد تشدید پس از LASIK: انجام PTK امکانپذیر است، اما اثربخشی پس از برداشتن فلپ LASIK بیشتر است1,8)
مراقبت پس از عمل: پس از PTK، قطره آنتیبیوتیک (لووفلوکساسین 0.5%) و قطره کورتیکواستروئید (فلوئورومتولون 0.1%) تا زمان بهبودی اپیتلیوم 4 بار در روز و سپس با کاهش دوز استفاده میشود. بهبودی اپیتلیوم معمولاً 3 تا 5 روز طول میکشد
DALK روشی است که اندوتلیوم را حفظ میکند. با استفاده از تکنیک حباب بزرگ (big bubble technique)، استروما تا بالای غشای دسمه جدا و برداشته میشود و استرومای دهنده بخیه زده میشود. از آنجایی که خطر رد اندوتلیوم وجود ندارد، پیشآگهی طولانیمدت بهتر از PK است3). در بررسی Kitazawa و همکاران، دید 5 ساله پس از DALK برای دیستروفیهای قرنیه مرتبط با TGFBI (شامل نوع دانهای و مشبک) به طور کلی خوب و میزان بقای پیوند بالا بود10). در ژاپن، این روش تحت پوشش بیمه درمانی قابل انجام است.
در دیستروفی گرانولار قرنیه (GCD)، تمامی روشهای LASIK، LASEK، PRK و SMILE منع مصرف دارند. پس از جراحی، کدورت قرنیه به سرعت تشدید شده و منجر به کاهش شدید بینایی میشود 1,8,9). پس از LASIK، رسوبات ریز دانهای متعددی بین فلپ و استرومای قرنیه تشکیل میشود. تشدید بیماری پس از LASIK شدیدتر از PRK بوده و حدت بینایی نهایی نیز بدتر است 1,8). گزارشهای موردی از کره و ژاپن موارد متعددی را توصیف کردهاند که در آنها بیماران بدون علامت، چند ماه تا چند سال پس از LASIK دچار کدورت قابل توجه قرنیه شده و نیاز به PTK یا پیوند قرنیه پیدا کردهاند 8,9).
Qاگر پس از انجام LASIK مشخص شود که فرد مبتلا به GCD است، چه اتفاقی میافتد؟
A
در موارد بروز GCD پس از LASIK، رسوبات دانهای به سرعت بین فلپ و استرومای قرنیه تشکیل میشوند. گزینههای درمانی شامل PTK پس از برداشتن فلپ LASIK، DALK و PK است که به ترتیب بررسی میشوند. مراجعه زودهنگام به متخصص چشم اهمیت دارد.
ژن TGFBI پروتئین ماتریکس خارج سلولی به نام TGFBIp (کراتواپیتلین، ۶۸ کیلودالتون) را کد میکند. TGFBIp در چسبندگی، مهاجرت و تکثیر سلولی نقش داشته و در استرومای طبیعی قرنیه نیز بیان میشود 1,5,7). هنگامی که جهش در ژن TGFBI رخ میدهد، TGFBIp جهشیافته نسبت به تجزیه پروتئینی حساسیت کمتری پیدا کرده و به صورت رسوبات نامحلول در استرومای قرنیه تجمع مییابد 5,7).
GCD1 ناشی از جهش Arg555Trp (R555W) است. TGFBIp جهشیافته به صورت هیالین (ماده شیشهای) در لایههای سطحی استرومای قرنیه رسوب میکند. آمیلوئید در این نوع وجود ندارد 3).
GCD2 تقریباً منحصراً ناشی از جهش Arg124His (R124H) است 1,5). در GCD2، هر دو ماده هیالین و آمیلوئید رسوب میکنند.
اختلال در اتوفاژی: در GCD2 اختلال در اتوفاژی گزارش شده است که منجر به کاهش تجزیه TGFBIp و افزایش تجمع آن میشود 1,5)
اختلال عملکرد میتوکندری: پیشنهاد شده است که TGFBIp جهشیافته خود بر فیبروبلاستهای قرنیه تأثیر گذاشته و ممکن است باعث اختلال عملکرد میتوکندری شود 1)
تأثیر عروق جدید قرنیه: در نواحی همراه با عروق جدید قرنیه، رسوبات تمایل به کاهش و جذب مجدد دارند. این یافته از مکانیسم تمرکز رسوبات در مرکز قرنیه که فاقد عروق خونی است، پشتیبانی میکند1)
پس از LASIK، TGFBIp به سرعت بین فلپ و بستر استروما رسوب میکند. تصور میشود این به دلیل دستکاری جراحی در مرکز قرنیه است که تجمع TGFBIp جهشیافته را تسریع میکند1,8). از آنجایی که برش قرنیه در جراحی آب مروارید (نزدیک لیمبوس) باعث تشدید نمیشود، فاصله از لیمبوس عروقی شده مرتبط فرض میشود1). مشاهدات پاتولوژیک Awwad و همکاران نشان میدهد که رسوبات تشکیلشده پس از LASIK همراه با فعال شدن کراتوسیتها در پاسخ به ترمیم زخم در سطح مشترک فلپ و بستر استروما، TGFBIp را تجمع میدهند8).
رسوبات GCD1 در میکروسکوپ نوری به عنوان مواد اسیدوفیل همگن مشاهده میشوند و با رنگآمیزی تریکروم ماسون قرمز رنگ میشوند. در سطح میکروسکوپ الکترونی، به صورت ساختارهای میلهای یا ذوزنقهای با چگالی الکترونی بالا به قطر 100 تا 500 نانومتر دیده میشوند6).
در GCD2، علاوه بر رسوبات هیالین، فیبریلهای آمیلوئید (قطر 8 تا 10 نانومتر) نیز مشاهده میشوند. فیبریلهای آمیلوئید با رنگآمیزی کنگو رد به رنگ نارنجی-قرمز رنگ میشوند و در میکروسکوپ پلاریزاسیون دوشکستی سبز سیبی نشان میدهند6). ویژگی دوگانه رنگآمیزی برای تشخیص قطعی پاتولوژیک GCD2 مفید است.
بر اساس آنالیز پروتئومیکس Poulsen و همکاران، در قرنیه بیماران GCD2، TGFBIp جهشیافته R124H در مقایسه با TGFBIp طبیعی کمتر توسط آنزیمهای پروتئولیتیک بریده میشود و قطعات خاص C ترمینال به طور انتخابی تجمع مییابند6). این مقاومت به برش ممکن است زمینهساز تشکیل فیبریلهای هیالین و آمیلوئید باشد.
گزارش شده است که کلرید لیتیوم تولید پروتئین TGFBI را کاهش میدهد. درمان ترکیبی ملاتونین و راپامایسین ممکن است بیان پروتئین TGFBI را مهار کرده و همزمان با فعال کردن اتوفاژی، تجزیه TGFBIp جهشیافته را تسریع کند1,5).
خاموش کردن بیان TGFBI جهشیافته با استفاده از RNA مداخلهگر کوچک (siRNA) یا RNA سنجاق سری کوتاه (shRNA) در مرحله پیشبالینی در حال مطالعه است. فناوری ویرایش ژنوم CRISPR/Cas9 نیز یک گزینه است، اما اثرات خارج از هدف ناخواسته بر آلل طبیعی یا سایر ژنها یک چالش است1,5).
الکترولیز قرنیه (corneal electrolysis): استفاده آزمایشی برای موارد عود پس از پیوند قرنیه گزارش شده است. نتایج طولانیمدت نامشخص است.
تشخیص به کمک یادگیری ماشین: توسعه مدل هوش مصنوعی برای شناسایی خودکار GCD از عکسهای بخش قدامی چشم گزارش شده است.
شاپرون درمانی: تحقیقات پایهای بر روی شاپرونهای شیمیایی (مانند 4-phenylbutyric acid) که به تاخوردگی صحیح TGFBIp جهشیافته کمک میکنند، در حال انجام است3).
لایه اپیتلیال قرنیه مشتق از سلولهای iPS: درمان پیوند لایه اپیتلیال قرنیه ساخته شده از سلولهای iPS بیمار به عنوان یک گزینه آینده در حال تحقیق است.
در داخل کشور، بحثها برای ایجاد یک ثبت ملی برای دیستروفیهای قرنیه مرتبط با TGFBI، به رهبری انجمن چشمپزشکی ژاپن و انجمن قرنیه ژاپن، ادامه دارد. با پوشش بیمهای آزمایش ژنتیک در سال 2020، موارد تشخیص ژنتیکی افزایش یافته است و دادههای پیگیری طولانیمدت از الگوهای جهش و فنوتیپهای خاص ژاپنی در حال جمعآوری است3,11). انتظار میرود در آینده، یک سیستم پزشکی مبتنی بر شواهد از غربالگری ناقلان تا تصمیمگیری برای جراحی انکساری ایجاد شود.
Chang MS, Jun I, Kim EK. Mini-Review: Clinical Features and Management of Granular Corneal Dystrophy Type 2. Korean journal of ophthalmology : KJO. 2023;37(4):340-347. doi:10.3341/kjo.2023.0032. PMID:37336511; PMCID:PMC10427907.
Weiss JS, Møller HU, Aldave AJ, et al. IC3D classification of corneal dystrophies-Edition 2. Cornea. 2015;34(2):117-159. doi:10.1097/ICO.0000000000000307. PMID:25564336.
Lisch W, Weiss JS. Clinical and genetic update of corneal dystrophies. Experimental eye research. 2019;186:107715. doi:10.1016/j.exer.2019.107715. PMID:31301286.
Han KE, Choi SI, Kim TI, Maeng YS, Stulting RD, Ji YW, et al. Pathogenesis and treatments of TGFBI corneal dystrophies. Progress in retinal and eye research. 2016;50:67-88. doi:10.1016/j.preteyeres.2015.11.002. PMID:26612778.
American Academy of Ophthalmology Cornea/External Disease Panel. Corneal Edema and Opacification Preferred Practice Pattern®. Ophthalmology. 2024;131(4):P1-P79.
Poulsen ET, Dyrlund TF, Runager K, et al. Proteomics of granular corneal dystrophy type 2 reveals altered TGFBIp proteolytic processing. J Proteome Res. 2014;13(10):4659-4667.
Dinh R, Rapuano CJ, Cohen EJ, Laibson PR. Recurrence of corneal dystrophy after excimer laser phototherapeutic keratectomy. Ophthalmology. 1999;106(8):1490-1497.
Awwad ST, Di Pascuale MA, Hogan RN, Forstot SL, McCulley JP, Cavanagh HD. Avellino corneal dystrophy worsening after laser in situ keratomileusis: further clinicopathologic observations and proposed pathogenesis. American journal of ophthalmology. 2008;145(4):656-61. doi:10.1016/j.ajo.2007.12.008. PMID:18243154; PMCID:PMC2661203.
Jun RM, Tchah H, Kim TI, et al. Avellino corneal dystrophy after LASIK. Ophthalmology. 2004;111(3):463-468.
Kitazawa K, Kawasaki S, Shinomiya K, et al. Safety of anterior lamellar keratoplasty for recurrent TGFBI corneal dystrophy. Br J Ophthalmol. 2016;100(4):448-454.